Hemes belong to a small subgroup of the tetrapyrrole family, which is characterized by the combination of a ferrous ion and a porphyrin macrocycle. The ‘true’ hemes possess a fully oxidized porphyrin macrocycle, including heme a, heme b, heme c, and heme o. Hemes are an important class of prosthetic molecules that play roles in a number of biological processes. The most widespread and ubiquitous is heme b, which plays an important role in transporting oxygen as part of hemoglobin. In addition, heme b is a cofactor for many enzymes, such as myoglobin, cytochrome P450, and peroxidases, and plays significant roles in catalysis, transcription, signaling, and electron transfer.
- heme b
- pathway
- protein
1. Introduction
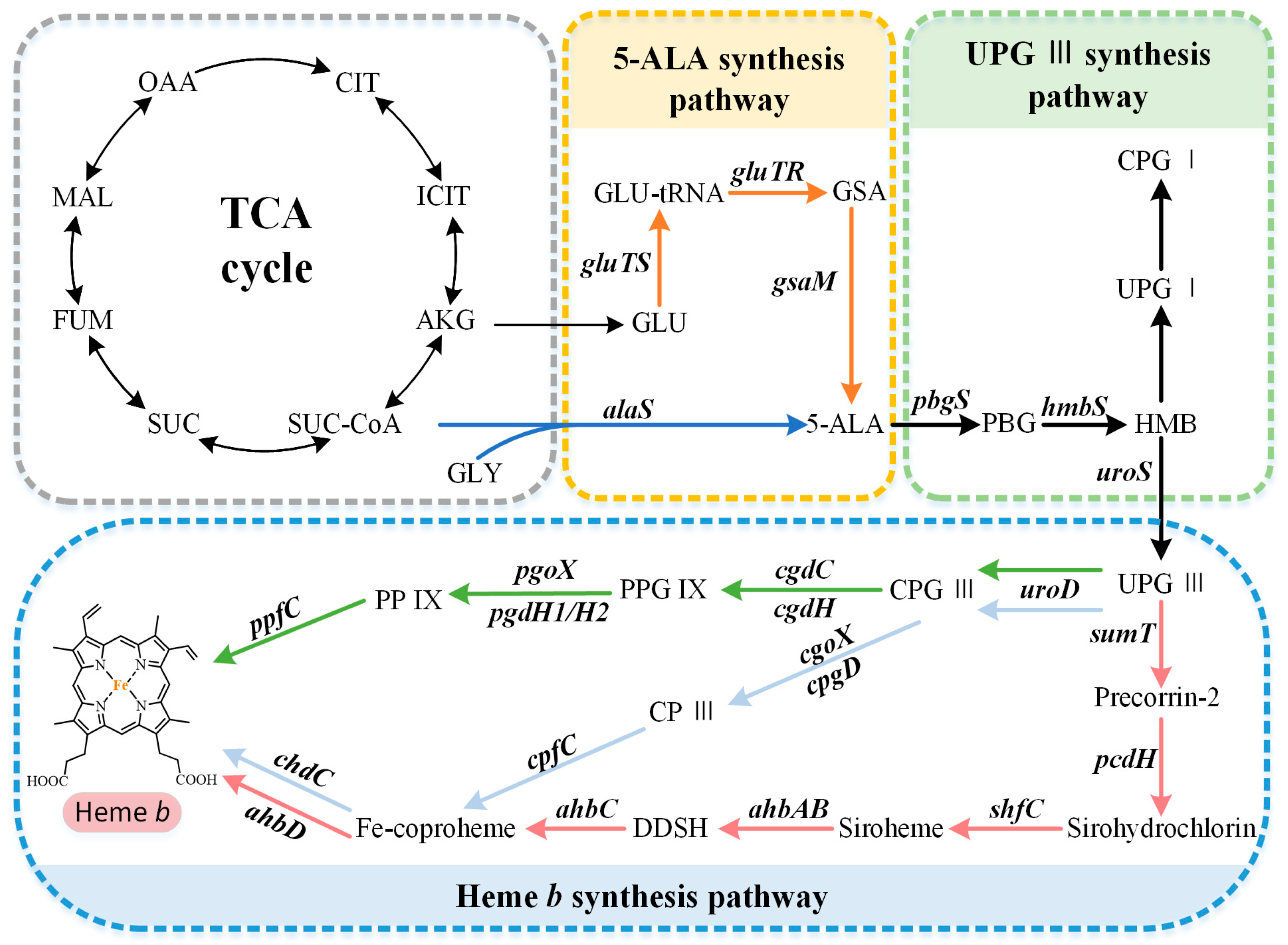
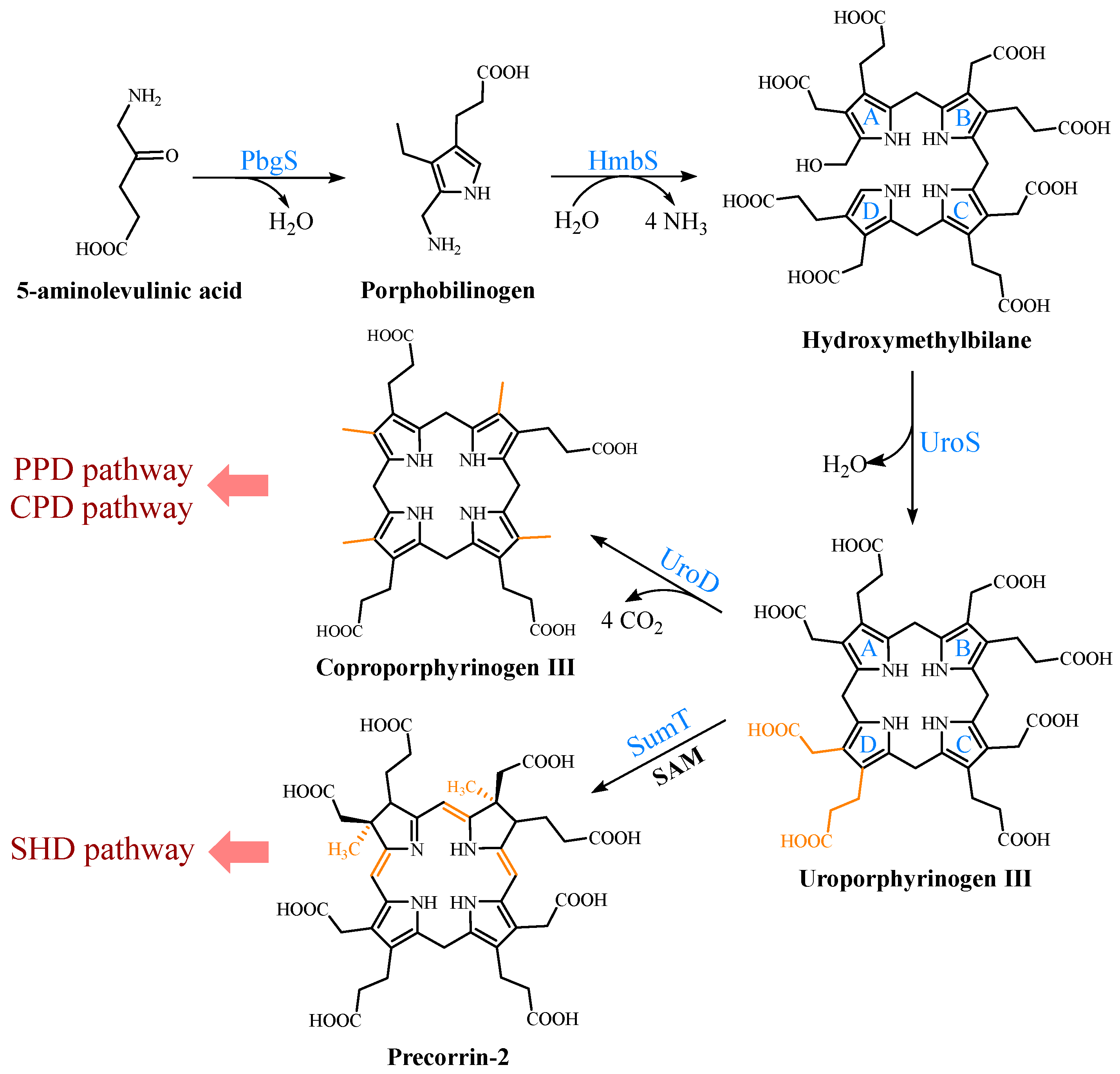
2. Biosynthetic Pathways of the Precursor 5-ALA
3. Formation of the Common Tetrapyrrole Core UPG III
4. Multiple Pathways for Synthesizing Heme b
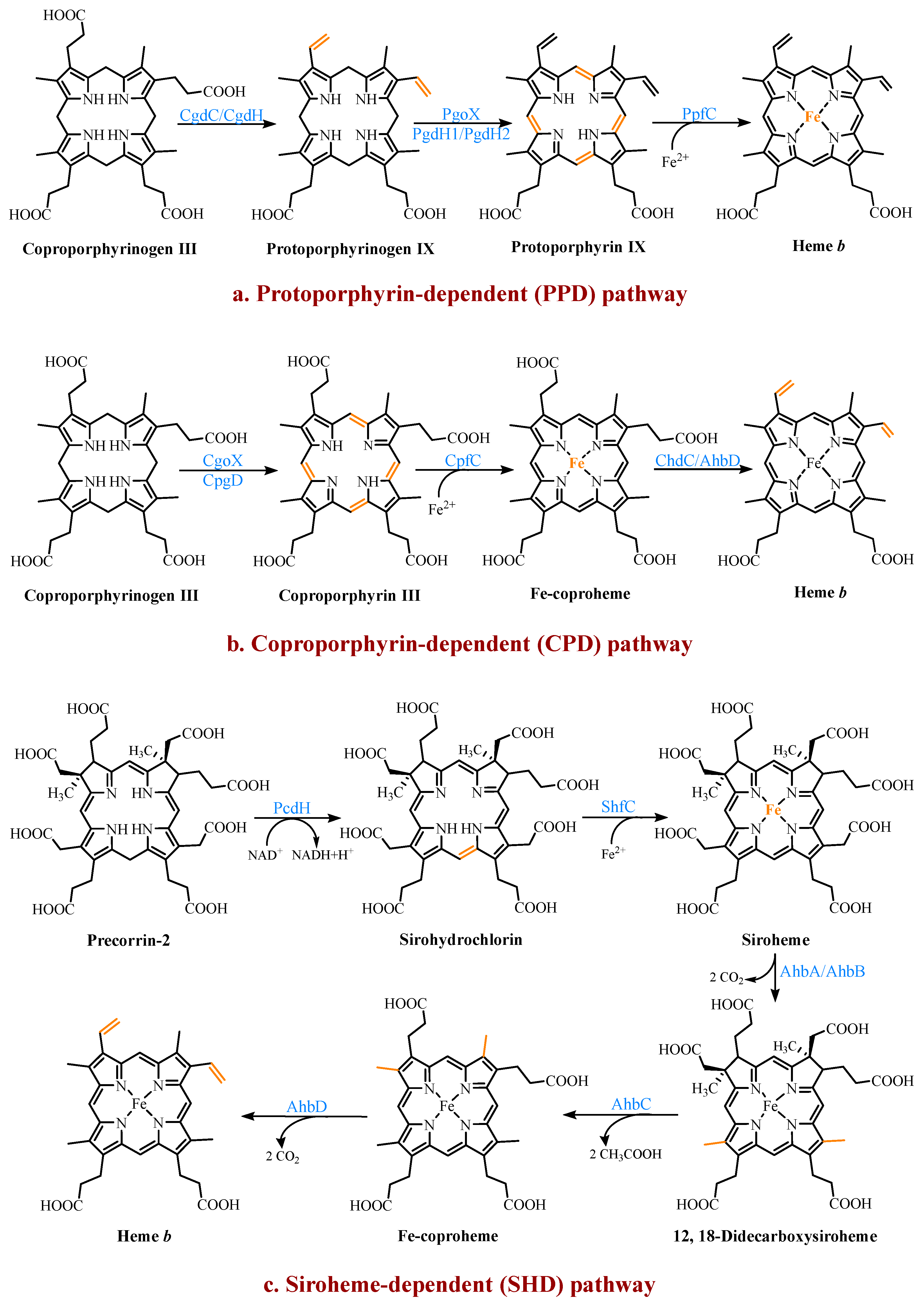
4.1. The Protoporphyrin-Dependent (PPD) Branch
4.2. The Coproporphyrin-Dependent (CPD) Branch
4.3. The Siroheme-Dependent (SHD) Pathway
This entry is adapted from the peer-reviewed paper 10.3390/molecules28083633
References
- Zhao, X.R.; Choi, K.R.; Lee, S.Y. Metabolic engineering of Escherichia coli for secretory production of free haem. Nat. Catal. 2018, 1, 720–728.
- Beale, S.I.; Castelfranco, P.A. 14C incorporation from exogenous compounds into δ-aminolevulinic acid by greening cucumber cotyledons. BBRC 1973, 52, 143–149.
- Kaufholz, A.L.; Hunter, G.A.; Ferreira, G.C.; Lendrihas, T.; Hering, V.; Layer, G.; Jahn, M.; Jahn, D. Aminolaevulinic acid synthase of Rhodobacter capsulatus: High-resolution kinetic investigation of the structural basis for substrate binding and catalysis. Biochem. J. 2013, 451, 205–216.
- Luer, C.; Schauer, S.; Virus, S.; Schubert, W.D.; Heinz, D.W.; Moser, J.; Jahn, D. Glutamate recognition and hydride transfer by Escherichia coli glutamyl-tRNA reductase. FEBS J. 2007, 274, 4609–4614.
- Ilag, L.L.; Jahn, D. Activity and spectroscopic properties of the Escherichia coli glutamate 1-semialdehyde aminotransferase and the putative active site mutant K265R. Biochemistry 1992, 31, 7143–7151.
- Nogaj, L.A.; Beale, S.I. Physical and kinetic interactions between glutamyl-tRNA reductase and glutamate-1-semialdehyde aminotransferase of Chlamydomonas reinhardtii. J. Biol. Chem. 2005, 280, 24301–24307.
- Zhang, J.; Cui, Z.; Zhu, Y.; Zhu, Z.; Qi, Q.; Wang, Q. Recent advances in microbial production of high-value compounds in the tetrapyrrole biosynthesis pathway. Biotechnol. Adv. 2022, 55, 107904.
- Breinig, S.; Kervinen, J.; Stith, L.; Wasson, A.S.; Fairman, R.; Wlodawer, A.; Zdanov, A.; Jaffe, E.K. Control of tetrapyrrole biosynthesis by alternate quaternary forms of porphobilinogen synthase. Nat. Struct. Biol. 2003, 10, 757–763.
- Coates, L.; Beaven, G.; Erskine, P.T.; Beale, S.I.; Wood, S.P.; Shoolingin-Jordan, P.M.; Cooper, J.B. Structure of Chlorobium vibrioforme 5-aminolaevulinic acid dehydratase complexed with a diacid inhibitor. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 1594–1598.
- Jaffe, E.K. The porphobilinogen synthase family of metalloenzymes. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 115–128.
- Jordan, P.M. The biosynthesis of 5-aminolaevulinic acid and its transformation into uroporphyrinogen III. New Compr. Biochem. 1991, 19, 1–66.
- Shoolingin-Jordan, P.M.; Warren, M.; Fau-Awan, S.J.; Awan, S.J. Discovery that the assembly of the dipyrromethane cofactor of porphobilinogen deaminase holoenzyme proceeds initially by the reaction of preuroporphyrinogen with the apoenzyme. Biochem. J. 1996, 316, 373–61996.
- Silva, P.J.; Ramos, M.J. Comparative density functional study of models for the reaction mechanism of uroporphyrinogen III synthase. J. Phys. Chem. B 2008, 112, 3144–3148.
- Lindsey, J.S.; Ptaszek, M.; Taniguchi, M. Simple formation of an abiotic porphyrinogen in aqueous solution. Orig. Life Evol. Biosph. 2009, 39, 495–515.
- Hashimoto, K.; Nakamura, K.; Kuroda, T.; Yabe, I.; Nakamatsu, T.; Kawasaki, H. The protein encoded by NCgl1221 in Corynebacterium glutamicum functions as a mechanosensitive channel. Biosci. Biotechnol. Biochem. 2010, 74, 2546–2549.
- Akhtar, M. Mechanism and stereochemistry of the enzymes involved in the conversion of uroporphyrinogen III into haem. New Compr. Biochem. 1991, 19, 67–99.
- Ishida, T.; Yu, L.; Akutsu, H.; Ozawa, K.; Kawanishi, S.; Seto, A.; Inubushi, T.; Sano, S. A primitive pathway of porphyrin biosynthesis and enzymology in Desulfovibrio vulgaris. Proc. Natl. Acad. Sci USA 1998, 95, 4853–4858.
- Bali, S.; Lawrence, A.D.; Lobo, S.A.; Saraiva, L.M.; Golding, B.T.; Palmer, D.J.; Howard, M.J.; Ferguson, S.J.; Warren, M.J. Molecular hijacking of siroheme for the synthesis of heme and d1 heme. Proc. Natl. Acad. Sci. USA 2011, 108, 18260–18265.
- Vévodová, J.; Graham, R.M.; Raux, E.; Schubert, H.L.; Roper, D.I.; Brindley, A.A.; Ian Scott, A.; Roessner, C.A.; Stamford, N.P.; Elizabeth Stroupe, M.; et al. Structure/function studies on a S-adenosyl-L-methionine-dependent uroporphyrinogen III methyltransferase (SUMT), a key regulatory enzyme of tetrapyrrole biosynthesis. J. Mol. Biol. 2004, 344, 419–433.
- Batlle, A.M.; Benson, A.; Rimington, C. Purification and properties of coproporphyrinogenase. Biochem. J. 1965, 97, 731–740.
- Cavallaro, G.; Decaria, L.; Rosato, A. Genome-based analysis of heme biosynthesis and uptake in prokaryotic systems. J. Proteome Res. 2008, 7, 4946–4954.
- Layer, G.; Reichelt, J.; Jahn, D.; Heinz, D.W. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010, 19, 1137–1161.
- Rand, K.; Noll, C.; Schiebel, H.M.; Kemken, D.; Dülcks, T.; Kalesse, M.; Heinz, D.W.; Layer, G. The oxygen-independent coproporphyrinogen III oxidase HemN utilizes harderoporphyrinogen as a reaction intermediate during conversion of coproporphyrinogen III to protoporphyrinogen IX. Biol. Chem. 2010, 391, 55–63.
- Qin, X.; Sun, L.; Wen, X.; Yang, X.; Tan, Y.; Jin, H.; Cao, Q.; Zhou, W.; Xi, Z.; Shen, Y. Structural insight into unique properties of protoporphyrinogen oxidase from Bacillus subtilis. J. Struct. Biol. 2010, 170, 76–82.
- Möbius, K.; Arias-Cartin, R.; Breckau, D.; Hännig, A.L.; Riedmann, K.; Biedendieck, R.; Schröder, S.; Becher, D.; Magalon, A.; Moser, J.; et al. Heme biosynthesis is coupled to electron transport chains for energy generation. Proc. Natl. Acad. Sci. USA 2010, 107, 10436–10441.
- Kobayashi, K.; Masuda, T.; Tajima, N.; Wada, H.; Sato, N. Molecular phylogeny and intricate evolutionary history of the three isofunctional enzymes involved in the oxidation of protoporphyrinogen IX. Genome Biol. Evol. 2014, 6, 2141–2155.
- Kato, K.; Tanaka, R.; Sano, S.; Tanaka, A.; Hosaka, H. Identification of a gene essential for protoporphyrinogen IX oxidase activity in the cyanobacterium Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. USA 2010, 107, 16649–16654.
- Shepherd, M.; Dailey, T.A.; Dailey, H.A. A new class of -cluster-containing protoporphyrin (IX) ferrochelatases. Biochem. J. 2006, 397, 47–52.
- Moody, M.D.; Dailey, H.A. Ferric iron reductase of Rhodopseudomonas sphaeroides. J. Bacteriol. 1985, 163, 1120–1125.
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the mitochondrial heme metabolism complex. PLoS ONE 2015, 10, e0135896.
- Masoumi, A.; Heinemann, I.U.; Rohde, M.; Koch, M.; Jahn, M.; Jahn, D. Complex formation between protoporphyrinogen IX oxidase and ferrochelatase during haem biosynthesis in Thermosynechococcus elongatus. Microbiology 2008, 154, 3707–3714.
- Dailey, H.A.; Gerdes, S.; Dailey, T.A.; Burch, J.S.; Phillips, J.D. Noncanonical coproporphyrin-dependent bacterial heme biosynthesis pathway that does not use protoporphyrin. Proc. Natl. Acad. Sci. USA 2015, 112, 2210–2215.
- Al-Karadaghi, S.; Hansson, M.; Nikonov, S.; Jonsson, B.; Hederstedt, L. Crystal structure of ferrochelatase: The terminal enzyme in heme biosynthesis. Structure 1997, 5, 1501–1510.
- Medlock, A.; Swartz, L.; Dailey, T.A.; Dailey, H.A.; Lanzilotta, W.N. Substrate interactions with human ferrochelatase. Proc. Natl. Acad. Sci. USA 2007, 104, 1789–1793.
- Medlock, A.E.; Carter, M.; Dailey, T.A.; Dailey, H.A.; Lanzilotta, W.N. Product release rather than chelation determines metal specificity for ferrochelatase. J. Mol. Biol. 2009, 393, 308–319.
- Corrigall, A.V.; Siziba, K.B.; Maneli, M.H.; Shephard, E.G.; Ziman, M.; Dailey, T.A.; Kirsch, R.E.; Meissner, P.N. Purification of and kinetic studies on a cloned protoporphyrinogen oxidase from the aerobic bacterium Bacillus subtilis. Arch. Biochem. Biophys. 1998, 358, 251–256.
- Zappa, S.; Li, K.; Bauer, C.E. The tetrapyrrole biosynthetic pathway and its regulation in Rhodobacter capsulatus. Adv. Exp. Med. Biol. 2010, 675, 229–250.
- Bali, S.; Palmer, D.J.; Schroeder, S.; Ferguson, S.J.; Warren, M.J. Recent advances in the biosynthesis of modified tetrapyrroles: The discovery of an alternative pathway for the formation of heme and heme d 1. Cell Mol. Life Sci. 2014, 71, 2837–2863.
- Stroupe, M.E.; Leech, H.K.; Daniels, D.S.; Warren, M.J.; Getzoff, E.D. CysG structure reveals tetrapyrrole-binding features and novel regulation of siroheme biosynthesis. Nat. Struct. Biol. 2003, 10, 1064–1073.
- Smith, M.A.; King, P.J.; Grimm, B. Transient-State kinetic analysis of synechococcus glutamate 1-semialdehyde aminotransferase. Biochemistry 1998, 37, 319–329.