Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Traumatic brain injury (TBI) is one of the most prevalent causes of morbidity in the United States and is associated with numerous chronic sequelae long after the point of injury. One of the most common long-term complaints in patients with TBI is sleep dysfunction. It is reported that alterations in melatonin follow TBI and may be linked with various sleep and circadian disorders directly (via cellular signaling) or indirectly (via free radicals and inflammatory signaling).
- traumatic brain injury
- melatonin
- insomnia
- therapeutic
1. Introduction
Traumatic brain injury (TBI) is a prominent cause of morbidity and mortality around the world, with an estimated global incidence of nearly 1/100, as 69 million individuals sustain a TBI every year [1][2]. Patients with TBI experience a complex symptom constellation, varying widely between individuals and persisting for several years after the initial injury [3][4]. Disruption of normal sleep patterns following TBI of any severity is one of the most common complaints experienced in both acute and chronic recovery phases. Estimates for prevalence vary widely, ranging anywhere from 30% to 70% [5][6][7]. Sleep quality complaints with TBI correlate strongly with mood and pain domains. They are difficult to disentangle, as they independently associate with impaired cognition, pain sensitization, and mood disorders [8][9]; they also prolong recovery after injury [10]. Considering the high prevalence and health burden of sleep disorders following TBI, interventions that optimize sleep may provide one of the greatest opportunities to improve long-term outcomes in this population.
A suitable therapeutic intervention requires a balance of safety and efficacy, and it may be most effectively utilized with an understanding of how its mechanism of action addresses underlying injury pathophysiology. Despite growing insight into the importance of sleep dysfunction following CNS injury, detailed mechanisms for the cause of these disturbances remain largely unknown as they are likely multidimensional, depending on injury patterns and individualized comorbidities. Further, the mechanism of sleep dysfunction post-TBI may differ by chronicity since the injury. Acute parenchymal disruption has been shown to be caused by a mixture of the force-based primary injury and subsequent secondary injury, which includes resultant metabolic disruption, oxidative stress, and inflammation that may pervade long after the initial injury occurred, culminating in eventual neurodegeneration [11][12]. An ideal therapeutic might mitigate one or multiple causative injury mechanisms while simultaneously promoting a high safety and efficacy profile.
Melatonin has well-established properties as a potent antioxidant that also functions as a signaling hormone, regulating sleep and circadian physiology. For example, direct changes in melatonin production, receptor concentration, and circadian rhythm function have been repeatedly observed following TBI [13][14][15][16][17][18][19]. Disruptions of endogenous melatonin signaling after TBI may partly explain some of the pathological phenotypes related to sleep, inflammation, and hormonal function. As a therapeutic, exogenous melatonin has had several challenges. A host of negative or inconclusive clinical insomnia studies [20] conflict, in part, with supportive studies in comorbid sub-populations [21][22]. There are also some studies showing safety and efficacy in the use of melatonin for enhancing reproductive health and fertility [23][24], while some animal studies suggest there might be a risk in prenatal and childhood development due to hormone signals [25][26][27] (although no human evidence of developmental harm from melatonin is known to the authors of this paper). These data, combined with a lack of federal regulation and reports of inaccurate quantification and impurities during manufacture [28], have led to its removal from clinical guidelines and recommendations for the treatment of sleep disorders. Nonetheless, clinical use continues, and certain indications, such as the use of melatonin in shifting circadian timing (e.g., for jet lag), remain widely accepted [29] due to what appears to be an acceptable safety margin and side effect profile. Some argue that the increasingly prolific use in over-the-counter supplementation as a circadian clock-shifting stimulus and a sleep aid in the general population could be cause for concern in some susceptible populations [29].
2. Primary Injury
Proposed mechanisms underlying sleep dysfunction following TBI can be subdivided by chronicity and subsequent microscopic or macroscopic effects. Acute injury mechanisms implicate acceleration-deceleration (blast and/or coup-contrecoup), resulting in axonal shearing and diffuse interruption of affiliated functional networks, theoretically including those associated with wakefulness and sleep, as shown in Figure 1 [30]. Cranial surface morphology exerts traumatic action in areas of high shear stress, such as the sphenoid ridge, inferior frontal, anterior temporal, and basal forebrain regions. These areas are rich in axonal projections mediating sleep and wakefulness, such as those from the locus coeruleus (noradrenergic pathway), the suprachiasmatic nucleus (circadian rhythm disorders), posterior hypothalamus (orexin neurons), and tuberomammillary nucleus (histaminergic pathway) [30].
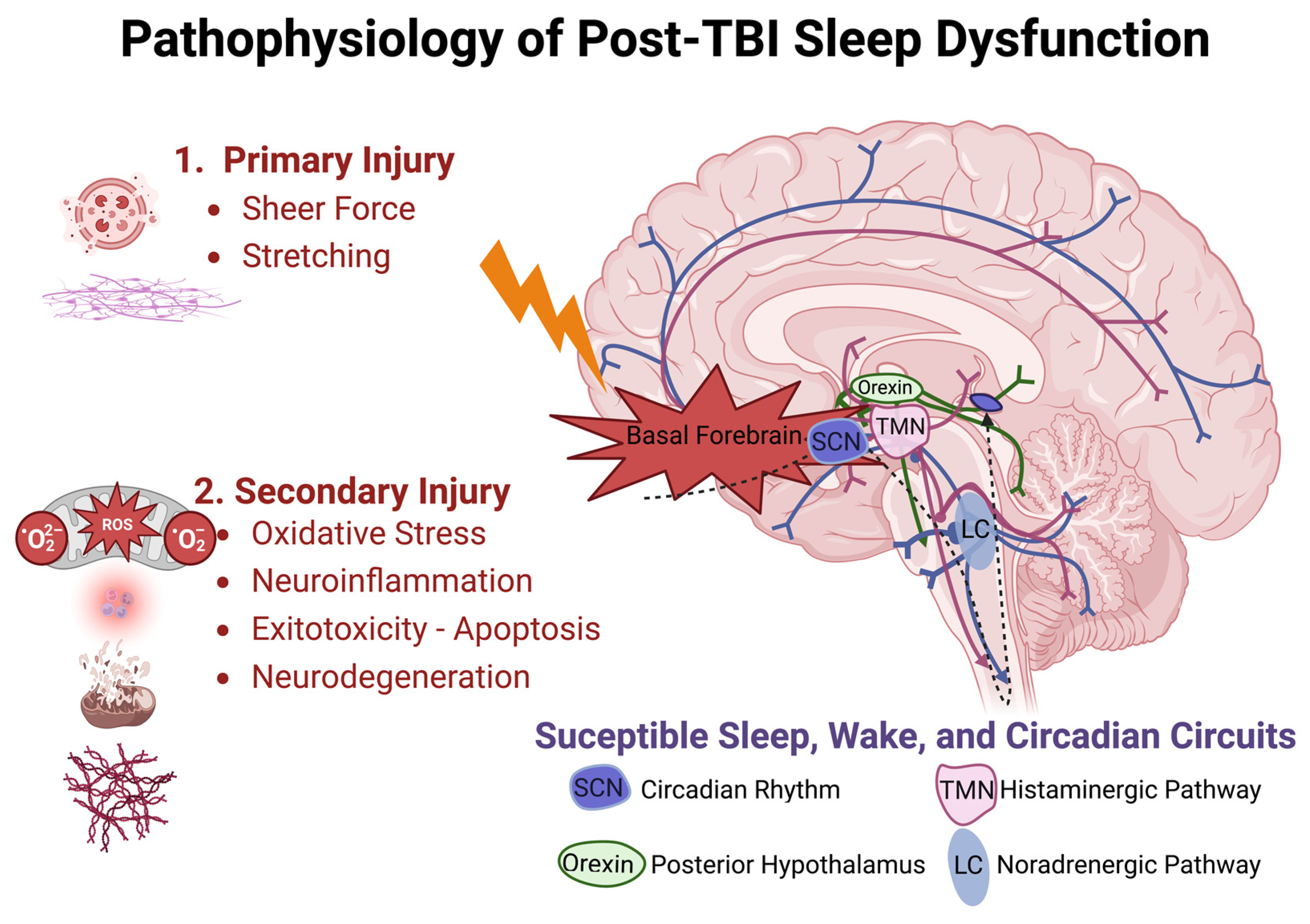
Figure 1. Pathophysiologic model of traumatic brain injury and disrupted sleep-related circuitry. Injury is sustained in two phases, with primary injury likely to disrupt axonal projections near the skull base and secondary injury responsible for prolonged cellular injury owed to oxidative stress, neuroinflammation, glutamate excitotoxicity, and neurodegeneration over acute and chronic durations. Suprachiasmatic Nucleus (SCN), Tuberomammillary Nucleus (TMN), Locus Coeruleus (LC).
Delayed mechanisms inciting injury include hypoxemia, hypotension, increased intracranial pressure, seizures, and hematoma formation. Microscopic effects of cellular damage, unchecked free radical production, neuroinflammation, and biochemical excitotoxicity-related events have all been shown to disrupt normal neural function following TBI.
3. Secondary Injury
Extensive inflammatory cytokine release is observed following TBI, functioning as an innate mechanism to promote self-healing and stabilize the parenchymal microenvironment of the CNS [31][32]. However, chronic inflammation can prolong clinical recovery and predispose patients to additional deficits [33][34]. Following primary injury, mediators of inflammatory cascades are released, which in turn promote the recruitment, activation, and integration of immune cells and signaling molecules within the cerebral microenvironment, as shown in Figure 2 [32]. The inflammatory response following primary injury is a prominent catalyst for secondary insults such as ischemia, edema, hemorrhage, lipid peroxidation/free radical injury, and cell death [34][35]. Secondary injuries can prolong treatment and impair a complete, timely recovery, representing an important focus for an interventional study.
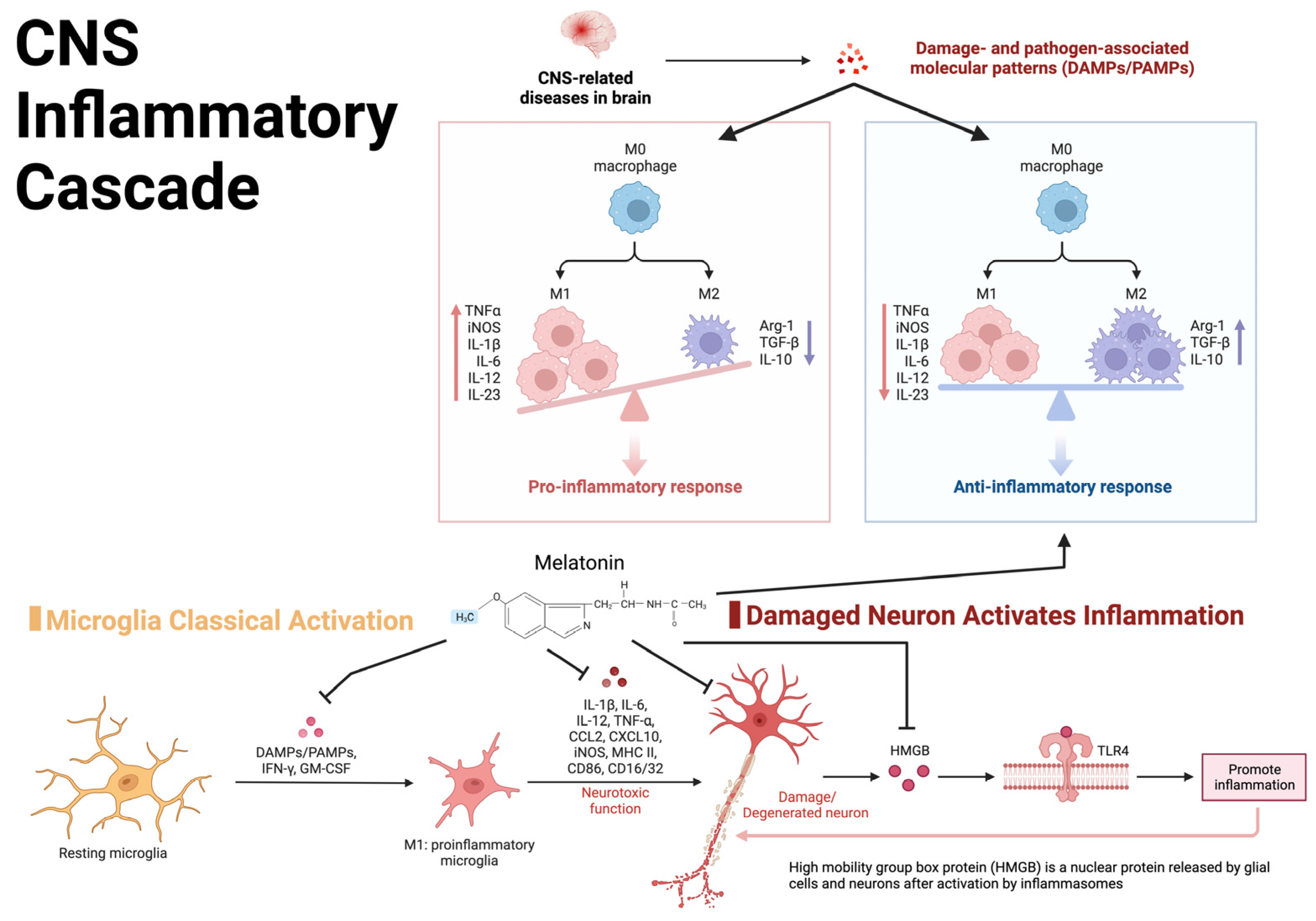
Figure 2. Visual representation of the central nervous system inflammatory cascade and the inflammatory feedback loop potentially leading to chronic post-TBI symptoms.
4. Injury Severity
The inciting force which produces clinically recognized TBI can be as innocuous as head jostling, common to many recreational sports, or as complex as an explosion resulting in multiple pressure waves, lacerations, contusions, and fragmented bones. Severe TBI has been found to result in loss of function in wake-promoting tuberomammillary histaminergic systems and is associated with symptoms of daytime fatigue. A post-mortem examination of patients with severe TBI found a loss of 41% of histaminergic neurons, 29% melanin-concentrating hormone, and 21% of orexinergic neurons [36]. Cortical excitability and stimulation likely underpin excessive daytime sleepiness (EDS) and fatigue in the TBI population. The magnitude of the force is not predictive of the severity of the injury. The severity of a TBI is dependent on a multitude of factors, including the mechanism of injury, characteristics of the individual patient, such as age, previous central nervous system (CNS) injury, and predisposing conditions [37].
5. Genetic Risk
Genomic variation may also expose individuals to the risk of developing sleep dysfunction following TBI. Genetic susceptibility for developing circadian rhythm disorders following TBI has been identified, providing additional context for mechanisms related to pathophysiology and risk factor stratification [38]. The PERIOD (Per) gene family, which is a polymorphic regulator of circadian rhythm, has been implicated in delayed sleep phase syndrome and confers increased risk for shorter sleep duration following TBI, as reported by Hong et al. [39]. Heterozygous Per3 carriers were associated with a significant risk for persistent sleep dysfunction following TBI [39]. Just as the magnitude of force does not always predict injury severity, TBI severity does not predict the severity of sleep-related symptoms; whether accounting for the genetic background would improve sleep outcome prediction remains an open question.
This entry is adapted from the peer-reviewed paper 10.3390/clockssleep5020016
References
- Feigin, V.L.; Theadom, A.; Barker-Collo, S.; Starkey, N.J.; McPherson, K.; Kahan, M.; Dowell, A.; Brown, P.; Parag, V.; Kydd, R.; et al. Incidence of traumatic brain injury in New Zealand: A population-based study. Lancet Neurol. 2013, 12, 53–64.
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.-C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2019, 130, 1080–1097.
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42.
- Kempf, J.; Werth, E.; Kaiser, P.R.; Bassetti, C.L.; Baumann, C.R. Sleep-wake disturbances 3 years after traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1402–1405.
- Ouellet, M.-C.; Beaulieu-Bonneau, S.; Morin, C.M. Sleep-wake disturbances after traumatic brain injury. Lancet Neurol. 2015, 14, 746–757.
- Mathias, J.; Alvaro, P. Prevalence of sleep disturbances, disorders, and problems following traumatic brain injury: A meta-analysis. Sleep Med. 2012, 13, 898–905.
- Sandsmark, D.K.; Elliott, J.; Lim, M.M. Sleep-Wake Disturbances After Traumatic Brain Injury: Synthesis of Human and Animal Studies. Sleep 2017, 40, zsx044.
- Fichtenberg, N.L.; Zafonte, R.D.; Putnam, S.; Mann, N.R.; Millard, A.E. Insomnia in a post-acute brain injury sample. Brain Inj. 2002, 16, 197–206.
- Gottshall, J.L.; Agyemang, A.A.; O’Neil, M.; Wei, G.; Presson, A.; Hewins, B.; Fisher, D.; Mithani, S.; Shahim, P.; Pugh, M.J.; et al. Sleep quality: A common thread linking depression, post-traumatic stress, and post-concussive symptoms to biomarkers of neurodegeneration following traumatic brain injury. Brain Inj. 2022, 36, 633–643.
- Wolfe, L.F.; Sahni, A.S.; Attarian, H. Sleep disorders in traumatic brain injury. Neurorehabilitation 2018, 43, 257–266.
- Mckee, A.C.; Daneshvar, D.H. The Neuropathology of Traumatic Brain Injury. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 127, pp. 45–66.
- Hemphill, M.A.; Dauth, S.; Yu, C.J.; Dabiri, B.E.; Parker, K.K. Traumatic Brain Injury and the Neuronal Microenvironment: A Potential Role for Neuropathological Mechanotransduction. Neuron 2015, 85, 1177–1192.
- Seifman, M.A.; Adamides, A.A.; Nguyen, P.N.; Vallance, A.S.; Cooper, D.J.; Kossmann, T.; Rosenfeld, J.V.; Morganti-Kossmann, M.C. Endogenous Melatonin Increases in Cerebrospinal Fluid of Patients after Severe Traumatic Brain Injury and Correlates with Oxidative Stress and Metabolic Disarray. J. Cereb. Blood Flow Metab. 2008, 28, 684–696.
- Marseglia, L.; D’Angelo, G.; Manti, S.; Rulli, I.; Salvo, V.; Buonocore, G.; Reiter, R.J.; Gitto, E. Melatonin Secretion Is Increased in Children with Severe Traumatic Brain Injury. Int. J. Mol. Sci. 2017, 18, 1053.
- Lorente, L.; Martín, M.M.; Abreu-González, P.; Pérez-Cejas, A.; Ramos, L.; Argueso, M.; Solé-Violán, J.; Cáceres, J.J.; Jiménez, A.; García-Marín, V. Serum melatonin levels in survivor and non-survivor patients with traumatic brain injury. BMC Neurol. 2017, 17, 138.
- Duclos, C.; Dumont, M.; Paquet, J.; Blais, H.; Van Der Maren, S.; Menon, D.K.; Bernard, F.; Gosselin, N. Sleep-wake disturbances in hospitalized patients with traumatic brain injury: Association with brain trauma but not with an abnormal melatonin circadian rhythm. Sleep 2019, 43, zsz191.
- Lorente, L.; Martín, M.M.; Ruiz, C.; Abreu-González, P.; Ramos-Gómez, L.; Argueso, M.; Sole-Violan, J.; Cáceres, J.J.; Jiménez, A. Serum melatonin levels in predicting mortality in patients with severe traumatic brain injury. Anaesth. Crit. Care Pain Med. 2021, 40, 100966.
- Osier, N.D.; Pham, L.; Pugh, B.J.; Puccio, A.; Ren, D.; Conley, Y.P.; Alexander, S.; Dixon, C.E. Brain injury results in lower levels of melatonin receptors subtypes MT1 and MT2. Neurosci. Lett. 2017, 650, 18–24.
- Rui, T.; Wang, H.; Li, Q.; Cheng, Y.; Gao, Y.; Fang, X.; Ma, X.; Chen, G.; Gao, C.; Gu, Z.; et al. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J. Pineal Res. 2020, 70, e12704.
- Choi, K.; Lee, Y.J.; Park, S.; Je, N.K.; Suh, H.S. Efficacy of melatonin for chronic insomnia: Systematic reviews and meta-analyses. Sleep Med. Rev. 2022, 66, 36179487.
- McGowan, N.M.; Kim, D.S.; Crespo, M.D.A.; Bisdounis, L.; Kyle, S.D.; Saunders, K.E.A. Hypnotic and Melatonin/Melatonin-Receptor Agonist Treatment in Bipolar Disorder: A Systematic Review and Meta-Analysis. CNS Drugs 2022, 36, 345–363.
- Marupuru, S.; Arku, D.; Campbell, A.M.; Slack, M.K.; Lee, J.K. Use of Melatonin and/on Ramelteon for the Treatment of Insomnia in Older Adults: A Systematic Review and Meta-Analysis. J. Clin. Med. 2022, 11, 5138.
- Hu, K.-L.; Ye, X.; Wang, S.; Zhang, D. Melatonin Application in Assisted Reproductive Technology: A Systematic Review and Meta-Analysis of Randomized Trials. Front. Endocrinol. 2020, 11, 160.
- Ghaderi, A.; Banafshe, H.R.; Mirhosseini, N.; Motmaen, M.; Mehrzad, F.; Bahmani, F.; Aghadavod, E.; Mansournia, M.A.; Reiter, R.J.; Karimi, M.-A.; et al. The effects of melatonin supplementation on mental health, metabolic and genetic profiles in patients under methadone maintenance treatment. Addict. Biol. 2018, 24, 754–764.
- González-Candia, A.; Veliz, M.; Araya, C.; Quezada, S.; Ebensperger, G.; Serón-Ferré, M.; Reyes, R.V.; Llanos, A.J.; Herrera, E.A. Potential adverse effects of antenatal melatonin as a treatment for intrauterine growth restriction: Findings in pregnant sheep. Am. J. Obstet. Gynecol. 2016, 215, 245.e1–245.e7.
- Colmenero, M.; Diaz, B.; Miguel, J.; González, M.; Esquifino, A.; Marin, B. Melatonin administration during pregnancy retards sexual maturation of female offspring in the rat. J. Pineal Res. 1991, 11, 23–27.
- Luboshitzky, R.; Shen-Orr, Z.; Nave, R.; Lavi, S.; Lavie, P. Melatonin administration alters semen quality in healthy men. J. Androl. 2002, 23, 572–578.
- Erland, L.; Saxena, P.K. Melatonin Natural Health Products and Supplements: Presence of Serotonin and Significant Variability of Melatonin Content. J. Clin. Sleep Med. 2017, 13, 275–281.
- Sun, S.-Y.; Chen, G.-H. Treatment of Circadian Rhythm Sleep–Wake Disorders. Curr. Neuropharmacol. 2021, 20, 1022–1034.
- Baumann, C.R.; Werner, J. TBI and Sleep–Wake Disorders: Pathophysiology, Clinical Management, and Moving towards the Future. Semin. Neurol. 2017, 37, 419–432.
- Xu, W.; Yue, S.; Wang, P.; Wen, B.; Zhang, X. Systemic inflammation in traumatic brain injury predicts poor cognitive function. Immun. Inflamm. Dis. 2021, 10, 8926513.
- Corps, K.N.; Roth, T.; McGAVERN, D.B. Inflammation and Neuroprotection in Traumatic Brain Injury. JAMA Neurol. 2015, 72, 355–362.
- Barlow, K.; Esser, M.M.J.; Veidt, M.; Boyd, R. Melatonin as a Treatment after Traumatic Brain Injury: A Systematic Review and Meta-Analysis of the Pre-Clinical and Clinical Literature. J. Neurotrauma 2019, 36, 523–537.
- Esposito, E.; Cuzzocrea, S. Antiinflammatory Activity of Melatonin in Central Nervous System. Curr. Neuropharmacol. 2010, 8, 228–242.
- Ikram, M.; Park, H.Y.; Ali, T.; Kim, M.O. Melatonin as a Potential Regulator of Oxidative Stress, and Neuroinflammation: Mechanisms and Implications for the Management of Brain Injury-Induced Neurodegeneration. J. Inflamm. Res. 2021, 14, 6251–6264.
- Valko, P.O.; Gavrilov, Y.V.; Yamamoto, M.; Finn, K.; Reddy, H.; Haybaeck, J.; Weis, S.; Scammell, T.E.; Baumann, C.R. Damage to histaminergic tuberomammillary neurons and other hypothalamic neurons with traumatic brain injury. Ann. Neurol. 2014, 77, 177–182.
- Prins, M.; Greco, T.; Alexander, D.; Giza, C.C. The pathophysiology of traumatic brain injury at a glance. Dis. Model. Mech. 2013, 6, 1307–1315.
- Schuch, J.B.; Genro, J.P.; Bastos, C.R.; Ghisleni, G.; Tovo-Rodrigues, L. The role of CLOCK gene in psychiatric disorders: Evidence from human and animal research. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2017, 177, 181–198.
- Hong, C.-T.; Wong, C.-S.; Ma, H.-P.; Wu, D.; Huang, Y.-H.; Wu, C.-C.; Lin, C.-M.; Su, Y.-K.; Liao, K.-H.; Ou, J.-C.; et al. PERIOD3 polymorphism is associated with sleep quality recovery after a mild traumatic brain injury. J. Neurol. Sci. 2015, 358, 385–389.
This entry is offline, you can click here to edit this entry!