Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Nuclei-based methods have become increasingly popular in the study of gene expression, epigenetics, and chromatin structure. To ensure the acquisition of biologically meaningful data, it is important to consider the available methodologies, future direction, and potential challenges and utilize improved designs and appropriate experimental strategies.
- nuclei isolation
- next-generation sequencing
- cell-type-specific isolation
- epigenetics
- transcriptomics
- single-cell sequencing
- single-nucleus sequencing
1. Introduction
The nucleus is the largest organelle in the eukaryotic cell, containing the genetic information (i.e., DNA) of a given organism [1]. In terms of its composition, the nucleus has its own double membrane nuclear envelope, which encapsulates the DNA-histone/DNA binding protein complexes (i.e., chromatin) in a well-organized state [2]. Within this state, the DNA forms a complex and high-order structure that ensures the efficient packing of the DNA in the nucleus and has a major effect on gene regulation, ultimately determining the given epigenetic and transcriptional profile of the cell. Due to the inherent properties and the content of the cellular nucleus, the number of studies that utilize nuclei for molecular investigations of cell-type-specific features has increased alongside the major research fields in molecular biology, such as genomics, transcriptomics and epigenomics. In fact, nuclei-based studies have come a long way since their initial discovery, exploration, isolation, and use as tools for molecular investigations [3][4][5].
In the last two decades, we have witnessed an upsurge in nuclei-based studies, particularly combined with high-throughput sequencing approaches. These studies aim to utilize isolated nuclei to identify both nucleus-associated properties and cell-type-specific features combined with supplementary techniques in molecular biology. The constant growth of next-generation sequencing (NGS), together with the steady reduction in sequencing cost, made the combination with nuclei-based studies both an affordable and increasingly powerful research tool [6]. Beyond this, the introduction of single-cell, as well as single-nucleus, sequencing technologies directed scientists to uncover the unique and heterogeneous molecular properties of a given cell population [7][8].
2. Nuclei-Isolation Procedures: From Cellular Dissociation to Nuclei Quality Check
2.1. Cellular Dissociation Methods
Nuclei isolation from tissues or cultured cells requires the dissociation of cells into single-cell suspensions [9][10]. Various cellular dissociation methods have been developed to isolate nuclei from different sources, such as enzymatic digestion, mechanical techniques, and combination approaches.
2.1.1. Enzymatic Digestion
Enzymatic digestion is one of the most commonly used methods for cellular dissociation. Enzymes such as trypsin, collagenase, papain and dispase can be used to break down cell–cell and cell–matrix interactions, releasing single cells [11][12][13][14] (Figure 1A). The strength of the enzyme needed for dissociation can vary depending on the type of tissue being dissociated. For instance, collagenase is commonly used for the dissociation of connective tissue, while trypsin is often used for the dissociation of epithelial cells [15]. Papain is another enzyme that is used for the dissociation of soft tissues [16]. Tissues that are difficult to dissociate, such as cartilage and bone, may necessitate stronger enzymes/reagents or suitable optimized protocols to achieve proper tissue dissociation and/or nuclear membrane permeabilization [16]. However, excessive exposure to enzymes can affect the viability and function of the cells, as well as the structure of nuclear components, which can affect downstream applications [17][18][19][20]. For instance, Denisenko and colleagues (2020) provided a systematic comparison of tissue dissociation protocols using both enzymatic digestions as well as nuclei-isolation approaches on adult mice kidneys, followed by single-cell and single-nuclei RNA-seq analyses [20]. The authors point out that enzymatic dissociation led to transcriptional changes consistent with a stress response, which was not observed with the non-enzymatic dissociation protocol. These observations are in agreement with previous studies reporting that enzymatic treatments could lead to major changes in cell-cycle status, induction of apoptosis, and structural alterations [18]. All these factors contribute to transcriptional changes that could lead to erroneous conclusions regarding the biological process under investigation [19]. Thus, it is important to optimize factors such as enzyme concentration, incubation time and temperatures to achieve efficient dissociation while maintaining the integrity of the nuclei.
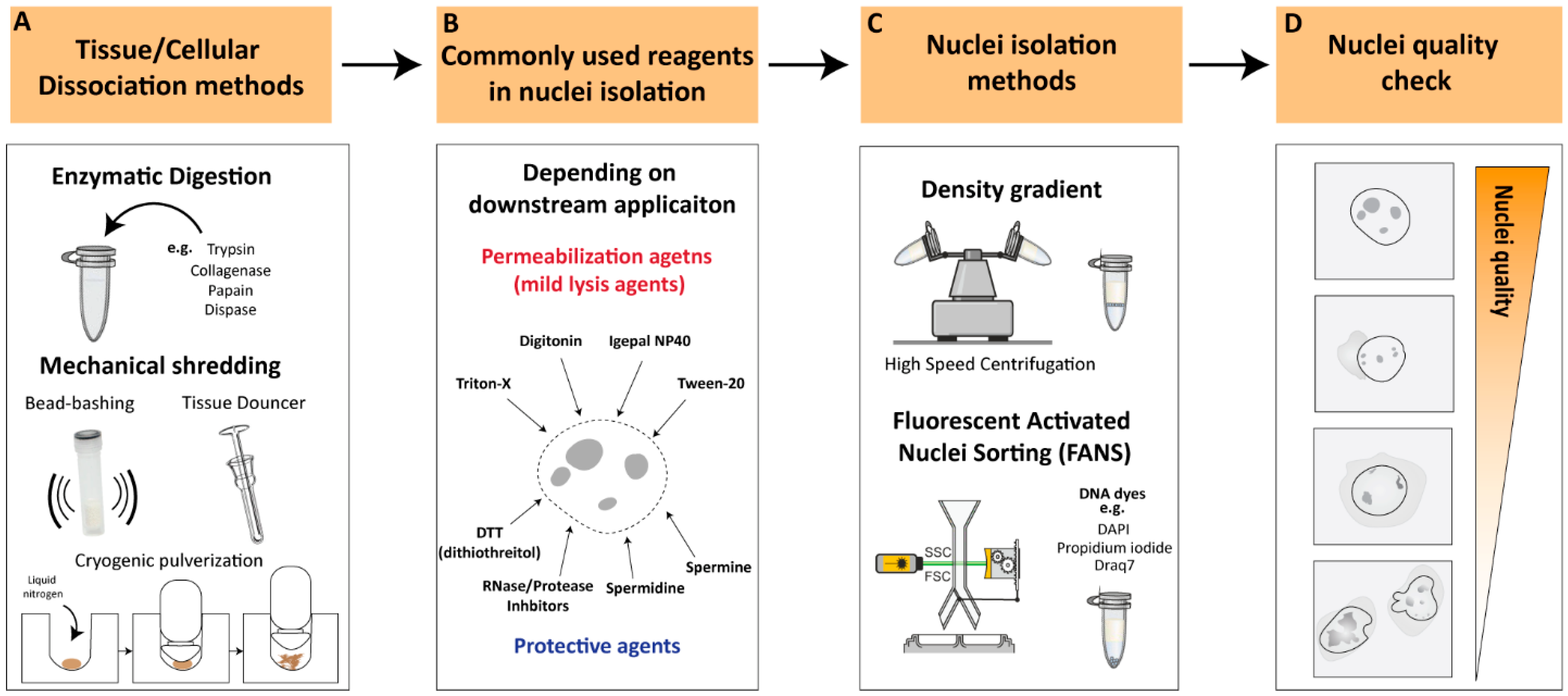
Figure 1. Key considerations for nuclei-isolation procedures (A). Tissue/cellular dissociation methods. Various cellular dissociation methods have been developed to isolate nuclei from different sources, such as enzymatic digestion, mechanical techniques and a combinatorial approach. (B). Reagents for nuclear permeabilization. Use of appropriate permeabilization buffers is crucial to ensure efficient and specific permeabilization of nuclei depending on downstream applications. (C). Nuclei-isolation methods. Density gradient centrifugations or flow-cytometry-based sorting are the most commonly used approaches for nuclei isolation. (D). Nuclei quality check. Illustration of high-quality nuclei are characterized by intact, round-shaped nuclei while low-quality nuclei often appear as small or fragmented particles with cytoplasmic debris or cellular aggregates. Low-quality nuclei could result in leakage of nuclear material from damaged or ruptured nuclei leading to contradictory observations.
2.1.2. Mechanical Dissociation
Additional commonly utilized methodologies for cellular dissociation involve mechanical dissociation and are often employed in combination with appropriate reagents to ensure proper tissue dissociation. Mechanical dissociation techniques such as Dounce homogenization, ‘bead-bashing’ and cryogenic pulverization are often used with nuclei-isolation methodologies (Figure 1A). For instance, Dounce homogenization involves grinding tissue against a tight-fitting pestle, and is commonly used in nuclei-isolation procedures, particularly from softer tissues such as brain [21]. Bead-bashing involves the mechanical agitation of cells using glass or plastic beads, which can physically break apart the cells and release nuclei. Bead-bashing is not typically utilized in conjunction with nuclear isolation methodologies, but it has demonstrated efficacy in dissociating various tissues, contingent upon the intensity of mechanical force applied and sample properties [22][23]. Cryogenic pulverization involves freezing tissue in liquid nitrogen and then pulverizing it into a fine powder and can be used for a wide range of tissues, including hard-to-dissociate tissues [24][25][26][27]. However, it should be noted that mechanical dissociation techniques can also damage nuclei and may require additional steps to optimize nuclear isolation and quality. Overall, the choice of cellular dissociation method depends on the source of the cells or tissues, the specific application, and the sensitivity of the nuclear components to the dissociation method. The careful optimization of dissociation protocols is crucial to ensure the efficient and specific isolation of nuclei while preserving the integrity of the nuclear components.
2.2. Nuclear Permeabilization and Protective Reagents
Nuclei permeabilization is a critical step in many nuclei-isolation procedures that involve the isolation and analysis of nuclear components. To permeabilize nuclei, various buffers or reagents are used to break down the nuclear membrane and allow access to nuclear contents. Commonly used buffers for nuclei permeabilization include mild lysis agents such as Triton X-100, Tween-20, digitonin and Igepal NP40 [27][28][29][30] (Figure 1B). These non-ionic detergents exhibit distinct chemical structures and properties that can solubilize the lipid bilayer of the nuclear membrane, allowing other molecules to enter the nucleus without disrupting the structure of the nuclear components. Often these detergents are used in combination with other detergents in relatively low concentrations (<0.5%) to improve their solubilizing properties, depending on the downstream application [28]. The selection of an optimal detergent is influenced by factors such as the source of the sample, input size, and downstream applications. The present entry will provide an overview of multiple investigations utilizing nuclei-isolation protocols, thereby allowing the reader to identify the most appropriate methodology for their specific research requirements.
To ensure efficient and high-quality nuclei without causing damage to nuclear components, it is recommended to supplement buffers with protective agents against nuclear degradation, depending on downstream applications (Figure 1B). Spermidine and spermine, which have antioxidant and stabilization functions on the chromatin structure, are often used as protective agents in nuclear isolation [27][31]. Another commonly employed protective reagent is DTT (dithiothreitol), a reducing agent utilized in nuclei-isolation protocols to preserve the integrity of the chromatin structure and promote the isolation of intact nuclei [17][20][27][30]. Additionally, RNAse and/or protease inhibitors are commonly added to the working buffer, depending on the downstream application, to prevent RNA and/or protein degradation during nuclear isolation processing. These reagents facilitate the isolation of intact nuclei, leading to the improved quality and yield of RNA samples for downstream analysis.
2.3. Nuclei-Isolation Methodologies
Nuclei-isolation procedures were established over half a century ago and involve density gradient centrifugation to visibly separate the nuclei from the rest of the raptured cell components [32][33][34]. Density gradient centrifugations rely on decreasing density solutions that amass the migrating target components according to their density during the centrifugation process [35]. The enveloped nuclei, containing the tightly packed DNA/proteins (i.e., highly dense structures), can be separated from the raptured and less-dense cytoplasmic compartment [36]. Density gradient nuclei-isolation approaches involve high-speed centrifugation, which results in a relatively intact, debris-free nuclei population and, therefore, is ideal as a rapid nuclei-isolation procedure (Figure 1C). However, nuclear integrity can greatly differ depending on the isolation method, nuclei storage buffer, and suspension time in intermediate buffer [37]. In recent years, an increasing number of studies have reported optimized nuclei-isolation procedures, which vary depending on the type of organism, tissue, or cell from which the nuclei were isolated (See ‘Critical considerations for efficient nuclei isolation from diverse biological sources’).
An additional commonly used methodology for nuclei isolation is flow-cytometry-based nuclei sorting [37][38]. The principle of flow cytometry is based on the ability of cells or nuclei to pass through a stream of fluid and be sorted according to size, granularity, and fluorescence properties (Figure 1C). In the case of nuclei isolation, flow cytometry can be used to sort and isolate nuclei based on their DNA content, which is typically measured by staining with fluorescent DNA dyes such as propidium iodide, Draq7 or 4′,6-diamidino-2-phenylindole (DAPI) [39][40]. Fluorescent DNA stains are often used in combination with FANS (Fluorescence-Activated Nuclei Sorting) to isolate nuclei based on their fluorescence signal and size, allowing for the separation of intact nuclei from other cellular components and debris [37]. FANS can be used to isolate nuclei from various sources, including tissues and cultured cells, and it has several advantages over other methods, such as the ability to sort nuclei based on their DNA content and the ability to sort multiple populations of nuclei simultaneously [21]. However, flow cytometry also has some limitations, such as the need for a specialized instrument, the requirement for a large number of cells/nuclei, and the potential for nuclei damage due to shear forces generated during the sorting process.
Overall, flow cytometry is a useful and powerful tool for nuclei isolation that can be used in conjunction with downstream applications such as genomics, transcriptomics, and epigenomics.
2.4. Nuclei Quality Check
Nuclei quality checking is a critical stage for evaluating and ensuring the integrity and purity of nuclei prior to their use in downstream applications, such as single-cell or genome-wide sequencing assays. The quality check of isolated nuclei can be conducted during or after the isolation procedure by using several methods. One way to assess nuclei quality is by using fluorescent dyes that specifically label the DNA or RNA of the nuclei, such as DAPI, and assess the nuclear integrity through microscopic examination or flow cytometry analysis, where the nuclei are identified based on their size and DNA content. High-quality nuclei can be characterized by intact, round-shaped nuclei with minimal damage or debris from other cellular components (Figure 1D). On the other hand, low-quality nuclei often appear as small or fragmented particles under the microscope and may contain cytoplasmic debris or cellular aggregates. Low-quality nuclei could result in the leakage of nuclear material from damaged or ruptured nuclei, leading to the contamination of cytoplasmic RNA, proteins, and other cellular components, potentially confounding downstream applications (Figure 1D). Thus, it is essential to perform quality control measures to ensure that the isolated nuclei are of high quality before proceeding with downstream applications.
2.5. Critical Considerations for Efficient Nuclei Isolation from Diverse Biological Sources
2.5.1. Nuclei Isolation from Distinct Organisms
With the increased use of genome-wide sequencing technologies, the interest in the isolation of single, separated nuclei has increased accordingly. Such an interest is extended to distinct organisms containing dissimilar genetic compositions. Early nuclei-isolation procedures, which involved density gradient centrifugations, focused mostly on the isolation of nuclei from mammalian tissues [33][35][41]. In fact, to this day most of the current studies that report optimized nuclei-isolation procedures are associated with mice or human dissected tissues [21][27][29][32][42]. However, an increasing number of studies report alternative nuclei isolation from distinct organisms, including invertebrate model organisms such as Drosophila melanogaster [43], Caenorhabditis elegans [44][45], non-model organisms such as diatoms [46] and a variety of plant species [47][48][49].
2.5.2. Nuclei Isolation from Distinct Tissues
Within an organism, distinct tissues comprise distinct cellular and physiological properties, which make their cellular isolation and processing challenging. For these reasons, the selection of nuclei versus whole-cell isolation procedures can vary according to the selected tissues. Previous studies addressed the potential rationale for the selection of isolation method according to the source of material, arguing that in contrary to nuclei-isolation procedures, whole-cell separation and isolation of single units often require enzymatic or substance treatments [9][10]. Remarkably, the understanding of nuclei versus whole-cell comparisons, including material isolation methods and transcriptional analyses, has been obtained recently from single-cell/nucleus comparison studies [50][51][52]. (Further information will follow later in this entry under the section ‘Single-cell and single-nucleus sequencing studies’). Furthermore, single-cell/-nucleus comparison studies stated that nuclei-isolation procedures are advantageous for certain hard-to-dissociate tissue types such as brain, bone, or adipose tissues [51][52]. This is probably due to the properties of the tissues where enzymatic dissociation would be inefficient, but the isolation of nuclei, which includes removing the external structural layers of the cell, would instead be very efficient. Therefore, tissue type will often dictate the selection of the isolation method, according to the experimental design and biological question investigated.
2.5.3. Cell-Type-Specific Nuclei Isolation
A key challenge in biology is to understand the origin and maintenance of cellular diversity within a single organism, despite the identical genetic composition. The interest in such phenomena led scientists to explore the molecular attributes of specific cell populations in order to identify their characteristics and varying roles within an organism. To further comprehend such cellular heterogeneity, several methodologies have been developed to isolate cell-type-specific populations based on their unique molecular features (e.g., specific gene and/or protein expression), thereby uncovering their unique genome-wide characteristics within a seemingly similar cell population. The use of nuclei has been demonstrated as a valuable tool, particularly when combined with genetic manipulation (e.g., Cre-loxP system) for cell-type-specific labelling [53]. For instance, the technique ‘Isolation of Nuclei in Tagged Cell Types’ (INTACT) is an affinity purification-based method that isolates genetically defined populations that express a fluorescently tagged nuclear membrane protein (e.g., Sun1GFP) [54][55]. While INTACT was initially developed in plants, its use has been adapted for other model organisms and has gradually evolved towards the analysis of multiple cell types within the mammalian system, primarily within the intensively investigated mammalian brain [37][43][56][57][58]. Another frequently used approach for cell-type-specific nuclei isolation is using flow-cytometry-based procedures using endogenous or exogenous fluorescence labeling approaches [59]. For instance, one frequently used method for neuronal isolation is using the neuronal nuclear marker NeuN [60], which has been used for a long time. It was not until 2012 that the terminology FANS was coined, emphasizing the requirement of nuclei-specific sorting procedures [61]. A detailed summary of the methodologies used to isolate cell-type-specific populations, including nuclei, and their association with genome-wide transcriptomic and epigenomic analyses is reviewed in [59]. Overall, nuclei-based approaches have entered the realms of day-to-day experimental routine, yet so far, with relatively little information about their distinct properties and differences compared to whole-cell studies.
3. Association of Nuclei with Next-Generation Sequencing
3.1. Genomics and Epigenomics
It has become clear that the nuclear content has a major role in cellular function and regulation. The more frequent utilization of nuclei has gone hand-in-hand with an elevated interest in genomics and epigenetics, as well as significant technological improvements in various high-throughput NGS platforms [62].
Such technologies have allowed for the detection of molecular changes on a genome-wide level, providing both comprehensive databases of specific cell identities as well as locus-specific alterations within a given experimental condition. The curiosity to uncover the internal function and content within the nuclei of distinct cellular populations led to the growth of the field of epigenetics and the development of experimental techniques dedicated to uncovering the multifaceted mechanisms of gene regulation. For instance, the mechanisms of DNA methylation and post-translational modification (PTMs) of histones rely on the deposition of chemical modifications on the DNA or DNA-associated proteins (e.g., histones) within the nucleus [63]. These mechanisms control gene expression, which influences cell function, and ultimately give each cell type its unique epigenetic and transcriptomic identity. The isolation of nuclei has been shown to be sufficient for conducting a variety of epigenetic assays as many of these assays require access to the contents within the nuclei. For example, techniques such as ChIP-seq [64], Hi-C [65], and ATAC-seq [66] are designed to investigate the dynamic chromatin structure and chromatin-bound proteins that reside within the nucleus. Regardless of the specific downstream application of these assays, isolated nuclei have been demonstrated to be a suitable starting material for various epigenetic analyses. Given the growing interest in epigenetic regulatory mechanisms, particularly in the context of high-throughput sequencing, the use of nuclei has become an attractive option for these applications.
3.2. Transcriptomics
3.2.1. RNA-seq
The process of transcription is the first stage that transforms biological information (i.e., genetic code) into a translated outcome or product (i.e., protein). The field of transcriptomics aims to monitor and quantify the complete set of transcripts, including coding and non-coding RNAs, within a given cell at a given condition [67]. The investigation of the transcriptome is crucial for understanding the functional elements of the genome and their roles within a cell or tissue. RNA-seq enables the assessment of genome-wide transcriptional states and hence has become the primary methodology to investigate the transcriptome and all its variations. Since the advancement of the next-generation sequencing approaches, we have seen a dominant use of the RNA-seq, which keeps increasing over time.
3.2.2. Nuclear RNA-seq
On the other hand, nuclear RNA-seq (nucRNA-seq), which refers to the sequencing of RNA isolated from the cell nucleus, has not been used nearly as frequently, which might reflect the novelty of such a sequencing technique. If we contemplate the association between nuclear RNA and next-generation sequencing, the first evident description of nucRNA-seq was reported by Mitchel and colleagues (2012), who took the opportunity to compare the nuclear transcriptome of erythroid cells with RNA polymerase II (RNAPII) occupancy [68]. Already at that time, the authors reported a large fraction of unspliced transcripts, which was detected by nucRNA-seq, hence foreseeing some of the challenges that will be discussed below.
3.2.3. Association of nucRNA-seq in ‘Multi-Omics’ Studies
An increasing number of studies seek to combine multiple ‘omics’ approaches within a given experimental design to identify specific and shared molecular properties of a given cellular population [69][70]. A frequent example of such experiments is the association of RNA-seq with epigenetic methodologies such as chromatin accessibility or DNA methylation within a single experimental setup. Similarly, the application of nucRNA-seq has been applied as a ‘multi-omic’ approach. For instance, Chongtham and colleagues (2021) performed a literature review comparing the use of two nuclei sorting techniques, INTACT and FANS, including subsequent molecular analyses used in conjugation with the investigated techniques [37]. The authors observed that ~60% of the analyzed studies combined more than one sequencing technique. This illustrates the frequent association of various ‘omics’ approaches within the same study, particularly in nuclei-based research. Using these associations, scientists can obtain broader and more comprehensive insights into particular cellular mechanisms per given condition. Depending on the biological question investigated and distinct sample requirements, nuclei are often coupled with additional approaches such as FANS or INTACT to isolate specific populations and diversify their use in various sequencing methodologies. Overall, the rise of the mentioned sequencing techniques and their association with nuclei is undoubtedly a landmark in the fields of molecular genomics, transcriptomics and epigenomics, which have already shifted towards single-cell and, also, increasingly, single-nucleus sequencing.
3.3. Single-Cell and Single-Nucleus Sequencing Studies
Single-cell studies have taken on an essential role in biological research, providing unprecedented insights into the characteristics of individual cells within a population. One of the most frequently utilized approaches is single-cell RNA-seq (scRNA-seq), which reveals the transcriptome of individual cells and highlights the heterogeneity among seemingly identical cell populations. However, in parallel, studies utilizing single-nucleus RNA-seq (snRNA-seq) have gradually risen in number as well, and with them, the inevitable question: what are the differences between scRNA-seq and snRNA-seq?
The first application of snRNA-seq occurred one decade ago, providing the basis for further developments of snRNA-seq methodologies [71]. Grindberg and colleagues (2013) applied the first snRNA-seq to uncover the dynamic transcriptome of mouse neuronal progenitor cells [71]. The authors noted that single-nuclei sequencing provides a unique insight into the exploration of neuronal transcriptomes since it ‘avoids requiring isolation of single-cell suspensions, eliminating potential changes in gene expression due to enzymatic-cell dissociation methods’. In their analysis, the authors took the opportunity to compare bulk nuclei versus bulk cells as well as single nucleus versus single cell, opening the avenue to the exploration of nuclear transcriptomics at the single-cell level.
Several studies followed and investigated the comparison between single nuclei and single cells, providing extensive information about the experimental design of such experiments, including material collection/isolation (of cells and nuclei) as well as a comparison of downstream analytical tools and biological differences between these two compartments. Comparing single-nucleus and single-cell RNA-seq within a single experimental scheme has become frequent [51][52][72][73][74][75]. The fundamental differences in RNA composition between nuclei and whole cells are noteworthy, which inevitably led to studies that explore and uncover such differences in various biological systems. For instance, Lake and colleagues (2017) compared single-nucleus RNA sequencing (snRNA-seq) and single-cell RNA sequencing (scRNA-seq) to investigate differences in gene expression between individual nuclei and intact cells in the human brain. They found that snRNA-seq was able to detect unique features of gene expression that were not detected by scRNA-seq. In particular, snRNA-seq was more sensitive to detecting low abundance transcripts and transcripts that were restricted to certain cell types. However, snRNA-seq also showed a lower overall detection rate of transcripts compared to scRNA-seq [72]. Bakken and colleagues (2018) compared scRNA-seq and snRNA-seq from the mouse visual cortex [74]. The authors suggest that although the number of transcripts detected from scRNA-seq is higher than snRNA-seq, the latter can be similarly associated with neuronal cell types. Interestingly, the authors highlighted that the incorporation of introns was required for comparable clustering analysis between snRNA-seq and scRNA-seq. They speculated that this is due to the long genes known to be brain-specific and that this helps defining the neuronal population analyzed [74]. In another study, Slyper and colleagues (2020) provided a comprehensive overview covering various cancer cell types, distinct protocol strategies, tissue acquisition, and sequencing methodologies of scRNA-seq and snRNA-seq [51]. In this study, the authors remark about the choice of using either single-cell or single-nucleus RNA-seq, stating, ‘The choice between scRNA-Seq and snRNA-Seq is typically driven by sample availability, logistics, and biological question’. The advantages of snRNA-seq include the decoupling of sample acquirement and processing, high recovery from tissues difficult to be dissociated, and sample multiplexing within specific approaches such as Drop-seq [51][76][77]. Nevertheless, the authors highlighted the importance of testing several tissue-specific dissociation methods as the output can vary depending on the type of dissected tissue and the processing procedure. In a similar study, Ding and colleagues (2020) provided a systematic comparison of single-cell and single-nucleus RNA-seq, focusing on the isolation method, sequencing platform, and computational analyses [52]. Comparable with previous studies, the authors remarked that the selection of single nuclei can be an important strategy that could be directed towards complex tissue types showing reduced gene expression alterations, as compared to cellular dissociation methods. Overall, these studies highlight the importance of considering the strengths and limitations of different single-cell analysis techniques when investigating gene expression patterns in complex tissues.
This entry is adapted from the peer-reviewed paper 10.3390/cells12071051
References
- Devos, D.P.; Gräf, R.; Field, M.C. Evolution of the nucleus. Curr. Opin. Cell Biol. 2014, 28, 8–15.
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220.
- Pederson, T. The nucleus introduced. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–16.
- Mekhail, K.; Moazed, D. The nuclear envelope in genome organization, expression and stability. Nat. Rev. Mol. Cell Biol. 2010, 11, 317–328.
- Köhler, A.; Hurt, E. Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell Biol. 2007, 8, 761–773.
- Illumina. An Introduction to Next-Generation Sequencing Technology. Available online: http://www.illumina.com/technology/next-generation-sequencing.html (accessed on 3 August 2022).
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630.
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272.
- Tomlinson, M.J.; Tomlinson, S.; Yang, X.B.; Kirkham, J. Cell separation: Terminology and practical considerations. J. Tissue Eng. 2013, 4, 1–14.
- Almeida, M.; Garcia-Montero, A.C.; Orfao, A. Cell Purification: A New Challenge for Biobanks. Pathobiology 2015, 81, 261–275.
- Volovitz, I.; Shapira, N.; Ezer, H.; Gafni, A.; Lustgarten, M.; Alter, T.; Ben-Horin, I.; Barzilai, O.; Shahar, T.; Kanner, A.; et al. A non-aggressive, highly efficient, enzymatic method for dissociation of human brain-tumors and brain-tissues to viable single-cells. BMC Neurosci. 2016, 17, 30.
- Reichard, A.; Asosingh, K. Best Practices for Preparing a Single Cell Suspension from Solid Tissues for Flow Cytometry. Cytom. Part A 2019, 95, 219.
- Mendibil, U.; Ruiz-Hernandez, R.; Retegi-Carrion, S.; Garcia-Urquia, N.; Olalde-Graells, B.; Abarrategi, A. Tissue-Specific Decellularization Methods: Rationale and Strategies to Achieve Regenerative Compounds. Int. J. Mol. Sci. 2020, 21, 5447.
- Montanari, M.; Burattini, S.; Ciacci, C.; Ambrogini, P.; Carloni, S.; Balduini, W.; Lopez, D.; Panza, G.; Papa, S.; Canonico, B. Automated–Mechanical Procedure Compared to Gentle Enzy-matic Tissue Dissociation in Cell Function Studies. Biomolecules 2022, 12, 701.
- Miersch, C.; Stange, K.; Röntgen, M. Effects of trypsinization and of a combined trypsin, collagenase, and DNase digestion on liberation and in vitro function of satellite cells isolated from juvenile porcine muscles. Vitr. Cell. Dev. Biol. Animal 2018, 54, 406.
- Yousef, H.; Czupalla, C.J.; Lee, D.; Butcher, E.C.; Wyss-Coray, T. Papain-based Single Cell Isolation of Primary Murine Brain Endothelial Cells Using Flow Cytometry. Bio-Protocol 2018, 8, e3091.
- Gao, M.; Guo, P.; Liu, X.; Zhang, P.; He, Z.; Wen, L.; Liu, S.; Zhou, Z.; Zhu, W. Systematic study of single-cell isolation from musculoskeletal tissues for single-sell sequencing. BMC Mol. Cell Biol. 2022, 23, 32.
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14.
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160.
- Denisenko, E.; Guo, B.B.; Jones, M.; Hou, R.; Kock, L.; Lassmann, T.; Poppe, D.; Clement, O.; Simmons, R.K.; Lister, R.; et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020, 21, 130.
- Nott, A.; Schlachetzki, J.C.M.; Fixsen, B.R.; Glass, C.K. Nuclei isolation of multiple brain cell types for omics interrogation. Nat. Protoc. 2021, 16, 1629–1646.
- Wei, S.; Levy, B.; Hoffman, N.; Cujar, C.; Rodney-Sandy, R.; Wapner, R.; D’Alton, M.; Williams, Z. A rapid and simple bead-bashing-based method for genomic DNA extraction from mammalian tissue. Biotechniques 2020, 68, 240–244.
- Bead Beating Guide|MP Biomedicals. Available online: https://www.mpbio.com/bs/bead-beating-technology-explained (accessed on 12 October 2022).
- Givens, R.M.; Mesner, L.D.; Hamlin, J.L.; Buck, M.J.; Huberman, J.A. Integrity of chromatin and replicating DNA in nuclei released from fission yeast by semi-automated grinding in liquid nitrogen. BMC Res. Notes 2011, 4, 1–16.
- Zerpa-Catanho, D.; Zhang, X.; Song, J.; Hernandez, A.G.; Ming, R. Ultra-long DNA molecule isolation from plant nuclei for ultra-long read genome sequencing. STAR Protoc. 2021, 2, 1.
- Ayhan, F.; Douglas, C.; Lega, B.C.; Konopka, G. Nuclei isolation from surgically resected human hippocampus. STAR Protoc. 2021, 2, 100844.
- Loft, A.; Herzig, S.; Schmidt, S.F. Purification of GFP-tagged nuclei from frozen livers of INTACT mice for RNA- and ATAC-sequencing. STAR Protoc. 2021, 2, 100805.
- Linke, D. Detergents: An Overview. Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2009; Chapter 34; pp. 603–617.
- Krishnaswami, S.R.; Grindberg, R.V.; Novotny, M.; Venepally, P.; Lacar, B.; Bhutani, K.; Linker, S.B.; Pham, S.; Erwin, J.A.; Miller, J.A.; et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat. Protoc. 2016, 11, 499–524.
- Maitra, M.; Nagy, C.; Chawla, A.; Wang, Y.C.; Nascimento, C.; Suderman, M.; Théroux, J.F.; Mechawar, N.; Ragoussis, J.; Turecki, G. Extraction of nuclei from archived postmortem tissues for single-nucleus sequencing applications. Nat. Protoc. 2021, 16, 2788–2801.
- Khan, A.U.; Mei, Y.H.; Wilson, T. A proposed function for spermine and spermidine: Protection of replicating DNA against damage by singlet oxygen. Proc. Natl. Acad. Sci. USA 1992, 89, 11426–11427.
- Chauveau, J.; Moulé, Y.; Rouiller, C. Isolation of pure and unaltered liver nuclei morphology and biochemical composition. Exp. Cell Res. 1956, 11, 317–321.
- Hadjiolov, A.A.; Tencheva, Z.S.; Bojadjieva-Mikhailova, A.G. Isolation and some characteristics of cell nuclei from brain cortex of adult cat. J. Cell Biol. 1965, 26, 383–393.
- Blobel, G.; Potter, V.R. Nuclei from Rat Liver: Isolation Method That Combines Purity with High Yield. Science 1966, 154, 1662–1665.
- Katholnig, K.; Poglitsch, M.; Hengstschläger, M.; Weichhart, T. Lysis gradient centrifugation: A flexible method for the isolation of nuclei from primary cells. Methods Mol. Biol. 2015, 1228, 15–23.
- Kim, K.; Guck, J. The Relative Densities of Cytoplasm and Nuclear Compartments Are Robust against Strong Perturbation. Biophys. J. 2020, 119, 1946–1957.
- Chongtham, M.C.; Butto, T.; Mungikar, K.; Gerber, S.; Winter, J. INTACT vs. FANS for Cell-Type-Specific Nuclei Sorting: A Comprehensive Qualitative and Quantitative Comparison. Int. J. Mol. Sci. 2021, 22, 5335.
- Eremenko, E.; Golova, A.; Stein, D.; Einav, M.; Khrameeva, E.; Toiber, D. FACS-based isolation of fixed mouse neuronal nuclei for ATAC-seq and Hi-C. STAR Protoc. 2021, 2, 3.
- Ligasová, A.; Koberna, K. DNA Dyes—Highly Sensitive Reporters of Cell Quantification: Comparison with Other Cell Quantification Methods. Molecules 2021, 26, 5515.
- Orchard, P.; Manickam, N.; Ventresca, C.; Vadlamudi, S.; Varshney, A.; Rai, V.; Kaplan, J.; Lalancette, C.; Mohlke, K.L.; Gallagher, K.; et al. Human and rat skeletal muscle single-nuclei multi-omic integrative analyses nominate causal cell types, regulatory elements, and SNPs for complex traits. Genome Res. 2021, 31, 2258–2275.
- Lovtrup-Rein, H.; McEwen, B.S. Isolation and fractionation of rat brain nuclei. J. Cell Biol. 1966, 30, 405–415.
- Cui, M.; Wang, Z.; Chen, K.; Shah, A.M.; Tan, W.; Duan, L.; Sanchez-Ortiz, E.; Li, H.; Xu, L.; Liu, N.; et al. Dynamic Transcriptional Responses to Injury of Regenerative and Non-regenerative Cardiomyocytes Revealed by Single-Nucleus RNA Sequencing. Dev. Cell 2020, 53, 102–116.
- Gupta, V.; Lazzaro, B.P. A robust method to isolate Drosophila fat body nuclei for transcriptomic analysis. Fly 2022, 16, 62–67.
- Steiner, F.A.; Talbert, P.B.; Kasinathan, S.; Deal, R.B.; Henikoff, S. Cell-type-specific nuclei purification from whole animals for genome-wide expression and chromatin profiling. Genome Res. 2012, 22, 766–777.
- Han, M.; Wei, G.; McManus, C.E.; Hillier, L.W.; Reinke, V.; Isolated, C. elegans germ nuclei exhibit distinct genomic profiles of histone modification and gene expression. BMC Genom. 2019, 20, 500.
- Annunziata, R.; Balestra, C.; Marotta, P.; Ruggiero, A.; Manfellotto, F.; Benvenuto, G.; Biffali, E.; Ferrante, M.I. An optimised method for intact nuclei isolation from diatoms. Sci. Rep. 2021, 11, 1681.
- Folta, K.M.; Kaufman, L.S. Isolation of Arabidopsis nuclei and measurement of gene transcription rates using nuclear run-on assays. Nat. Protoc. 2006, 1, 3094–3100.
- Sikorskaite, S.; Rajamäki, M.L.; Baniulis, D.; Stanys, V.; Valkonen, J.P.T. Protocol: Optimised methodology for isolation of nuclei from leaves of species in the Solanaceae and Rosaceae families. Plant Methods 2013, 9, 31.
- Moro, B.; Kisielow, M.; Borrero, V.B.; Bouet, A.; Brosnan, C.A.; Bologna, N.G. Nuclear RNA purification by flow cytometry to study nuclear processes in plants. STAR Protoc. 2021, 2, 1.
- Wu, H.; Kirita, Y.; Donnelly, E.L.; Humphreys, B.D. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. J. Am. Soc. Nephrol. 2019, 30, 23–32.
- Slyper, M.; Porter, C.B.M.; Ashenberg, O.; Waldman, J.; Drokhlyansky, E.; Wakiro, I.; Smillie, C.; Smith-Rosario, G.; Wu, J.; Dionne, D.; et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020, 26, 792–802.
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T.; et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020, 38, 737–746.
- Tang J C, Y.; Rudolph, S.; Dhande O, S.; Abraira, V.E.; Choi, S.; Lapan, S.W.; Drew, I.R.; Drokhlyansky, E.; Huberman, A.D.; Regher, W.G. Cell type–specific manipulation with GFP-dependent Cre recombinase. Nat. Neurosci. 2015, 18, 1334–1341.
- Deal, R.B.; Henikoff, S. A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev. Cell 2010, 18, 1030–1040.
- Deal, R.B.; Henikoff, S. The INTACT method for cell typeg-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc. 2011, 6, 56–68.
- Henry, G.L.; Davis, F.P.; Picard, S.; Eddy, S.R. Cell type-specific genomics of Drosophila neurons. Nucleic Acids Res. 2012, 40, 9691–9704.
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 2015, 86, 1369–1384.
- Mo, A.; Luo, C.; Davis, F.P.; Mukamel, E.A.; Henry, G.L.; Nery, J.R.; Urich, M.A.; Picard, S.; Lister, R.; Eddy, S.R.; et al. Epigenomic landscapes of retinal rods and cones. eLife 2016, 5, e11613.
- Handley, A.; Schauer, T.; Ladurner, A.G.; Margulies, C.E. Designing Cell-Type-Specific Genome-wide Experiments. Mol. Cell 2015, 58, 621–631.
- Jiang, Y.; Matevossian, A.; Huang, H.S.; Straubhaar, J.; Akbarian, S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008, 9, 42.
- Haenni, S.; Ji, Z.; Hoque, M.; Rust, N.; Sharpe, H.; Eberhard, R.; Browne, C.; Hengartner, M.O.; Mellor, J.; Tian, B.; et al. Analysis of C. elegans intestinal gene expression and polyadenylation by fluorescence-activated nuclei sorting and 3′-end-seq. Nucleic Acids Res. 2012, 40, 6304–6318.
- Besser, J.; Carleton, H.A.; Gerner-Smidt, P.; Lindsey, R.L.; Trees, E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin. Microbiol. Infect. 2018, 24, 335–341.
- Wang, Y.C.; Peterson, S.E.; Loring, J.F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014, 2, 143–160.
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837.
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293.
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218.
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57.
- Mitchell J, A.; Clay, I.; Umlauf, D.; Chen, C.; Moir, C.A.; Eskiw, C.H.; Schoenfelder, S.; Chakalova, L.; Nagano, T.; Fraser, P. Nuclear RNA sequencing of the mouse erythroid cell transcriptome. PLoS ONE 2012, 7, e49274.
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83.
- Pinu, F.R.; Beale, D.J.; Paten, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D. Systems biology and multi-omics integration: Viewpoints from the metabolomics research community. Metabolites 2019, 9, 76.
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Araúzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-sequencing from single nuclei. Proc. Natl. Acad. Sci. USA 2013, 110, 19802–19807.
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2017, 36, 70–80.
- Lacar, B.; Linker, S.B.; Jaeger, B.N.; Krishnaswami, S.; Barron, J.; Kelder, M.; Parylak, S.; Paquola, A.; Venepally, P.; Novotny, M.; et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun. 2016, 7, 11022.
- Bakken, T.E.; Hodge, R.D.; Miller, J.A.; Yao, Z.; Nguyen, T.N.; Aevermann, B.; Barkan, E.; Bertagnolli, D.; Casper, T.; Dee, N.; et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE 2018, 13, e0209648.
- Korrapati, S.; Taukulis, I.; Olszewski, R.; Pyle, M.; Gu, S.; Singh, R.; Griffiths, C.; Martin, D.; Boger, E.; Morell, R.J.; et al. Single Cell and Single Nucleus RNA-Seq Reveal Cellular Heterogeneity and Homeostatic Regulatory Networks in Adult Mouse Stria Vascularis. Front. Mol. Neurosci. 2019, 12, 316.
- Kang, H.M.; Subramaniam, M.; Targ, S.; Nguyen, M.; Maliskova, L.; McCarthy, E.; Wan, E.; Wong, S.; Byrnes, L.; Lanata, C.M.; et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018, 36, 89–94.
- Stoeckius, M.; Zheng, S.; Houck-Loomis, B.; Hao, S.; Yeung, B.Z.; Mauck, W.M.; Smibert, P.; Satija, R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018, 19, 224.
This entry is offline, you can click here to edit this entry!