STING is an adjuvant protein on the endoplasmic reticulum (ER) which recognizes the cyclic dinucleotides (CDNs) generated by cyclic GMP-AMP synthase (cGAS) [
10]. cGAS can recognize not only foreign DNA, but also DNA from the cell itself [
11]. Once cGAS senses cytoplasmic double-stranded DNA (dsDNA), it converts GTP and ATP into 2′,3′-cGAMP, which binds and activates STING [
12], and it eventually secretes type I IFN and various proinflammatory factors [
13]. As part of the feedback loop of IFNs, both cGAS and STING expression can be upregulated positively by type I IFNs in order to amplify the immune response [
14,
15]. Meanwhile, STING and its upstream IFN-γ inducible protein 16 (IFI16) are also downregulated in order to prevent excessive inflammation [
16,
17]. The over-activation of STING is reported to be related to psoriasis, systemic lupus erythematosus (SLE), infectious diseases, non-alcoholic fatty liver disease (NAFLD), and other interferonopathies including STING-associated vasculopathy in infants (SAVI) and Aicardi–Goutières syndrome (AGS) [
13,
18,
19,
20]. Many STING inhibitors have been discovered, and their activities have been continuously improved through structural modifications. Li et al. isolated a STING inhibitor—Astin C—from the natural plant
Aster tataricus, which was reported to alleviate palmitic acid-induced cardiomyocyte contractile dysfunction [
21,
22]. C-176, the covalent STING inhibitor, showed remarkable efficacy in mouse models of diabetic cardiomyopathy and regulated pancreatic β-cell function [
23,
24]. Moreover, H-151, which was obtained through high-throughput screening after C-176, was tested with promising results in treating psoriasis [
25], myocardial infarction [
26], acute kidney injury [
27], and acute lung injury in mice [
28]. However, due to its relatively weak potency and poor pharmacokinetic properties, current STING inhibitors are still in the early stages, and there are no compounds being tested in clinical studies. Moreover, the high heterogeneity of the human STING (hSTING) gene also presents challenges to the development of STING inhibitors with clinical potential [
29].
2. The Structure and Location of STING
STING (also known as TMEM173, MITA, ERIS, and MPYS) [
30,
31,
32,
33,
34] is an ER membrane protein composed of 379 amino acids [
35]. The C-terminus domain (CTD) of STING contains a ligand-binding domain (LBD), and the N-terminus contains four transmembrane (TM) domains. The C-terminus also includes a dimerization domain and a C-terminal tail (CTT) domain containing the phosphorylation site of TANK-binding kinase 1 (TBK1) [
36]. The dimerization domain consists of hydrophobic amino acids that approach each other through hydrophobic interactions to form a V-shaped dimer where the binding site for cyclic dinucleotides (CDNs) is located [
37,
38,
39]. CDNs include cyclic diadenosine monophosphate (c-di-AMP), cyclic diguanylate (c-di-GMP), and cyclic GMP-AMP (cGAMP). In mammalian cells, the endogenous cGAMP includes two phosphodiester linkages: one between GMP 2′-OH and AMP 5′-phosphate and the other between AMP 3′-OH and GMP 5′-phosphate, which is called 2′,3′-cGAMP [
40,
41]. Compared with alternatively formed cGAMPs, 2′,3′-cGAMP showed the strongest affinity to hSTING, with a reported K
d value 300 times lower than that of c-di-GMP and 3′,3′-cGAMP, and 75 times lower than 2′,2′-cGAMP [
21,
40]. Additionally, the binding of c-di-GMP to STING is exothermic, while that of 2′,3′-cGAMP is endothermic [
40].
Based on the Human Protein Atlas (HPA) database [
42], STING expression has low tissue specificity and is expressed in almost all tissues. Except for B cells, STING is expressed in almost all immune cells. STING expression is higher in the lung, bronchi, tonsil, lymph node, and spleen, and lower in the skin, stomach, kidney, and adipose tissue.
3. The Function of STING
The cytoplasm of eukaryotic cells is devoid of DNA under normal physiological conditions, and any small amounts that may leak into the cytoplasm are swiftly degraded by nucleases [
43]. The detection of cytoplasmic DNA by the innate immune system induces various inflammatory responses and defense mechanisms [
39]. Since there is no DNA-binding domain in the STING structure, other pattern-recognition receptors are required in order to help recognize DNA, like cGAS. The threshold of STING activation is vital for the body to distinguish its own basic DNA level from abnormal conditions [
44]. Under unstimulated conditions, STING predominantly resides on the ER, although it has also been reported to be present on the mitochondria-associated ER membrane [
30]. Upon activation, STING traffics via the Golgi apparatus to form discrete punctate polymers around the nucleus [
30,
45]. DNA receptor cGAS can recognize abnormally exposed cytoplasmic DNA and synthesizes 2′,3′-cGAMP, including viral and bacterial DNA, DNA produced by the reverse transcription of RNA viruses, and DNA produced by cell damage [
45]. The binding of cGAMP to the STING dimer induces the rotation and closure of STING LBD and the release of STING CTT, leading to the formation of disulfide-linked polymers (
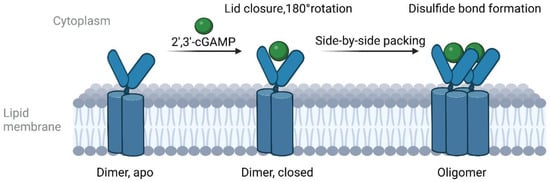
Figure 1) [
44,
46]. STING oligomerization occurs in the ER [
44]. After activation, STING is translocated from the ER via the ER–Golgi intermediate compartment (ERGIC) to the Golgi apparatus, where TBK1 and interferon regulatory factor 3 (IRF3) are recruited [
36,
47]. TBK1 phosphorylates STING and IRF3 after TBK1 autophosphorylation, inducing the dimerization of IRF3 [
36,
47]. IRF3 dimers are then transported into the nucleus and promote the expression of type I IFN and related immune factors. After activation, STING is transported to the lysosome for degradation [
48,
49].
Figure 1. Cartoon model of the cGAMP-induced oligomerization of wild-type STING.
The expression of inflammatory factors can be upregulated by activation of the NF-κB pathway [
50]. TRAF6, an E3 ubiquitin ligase of the TRAF family, is essential in the non-canonical NF-κB pathway during STING activation [
51,
52,
53,
54]. Abe et al. reported that STING and TBK1 could facilitate the dsDNA-induced activation of the NF-κB pathway with the assistance of TRAF6 [
52]. Additionally, Ataxia Telangiectasia mutated protein (ATM) and poly(ADP-ribose) polymerase 1 (PARP-1) can form a STING-activating complex with TRAF6, IFN-γ inducible protein 16 (IFI16), and p53 in response to DNA damage [
53]. Subsequently, TRAF6 catalyzes STING K63-linked ubiquitination in order to activate the NF-κB pathway independently of TBK1 [
53]. The special sequence of the zebrafish STING CTT can even recruit TRAF6 in order to boost NF-κB signaling [
54]. In contrast, TRAF6 and TBK1 play dispensable roles in the canonical NF-κB response induced by STING [
55]. These findings shed light on the intricate and diverse mechanisms underlying STING-mediated immune responses.
Recently, a novel discovery highlighted the STING-PKR-like ER kinase (PERK)-eukaryotic initiation factor-2α(eIF2α) pathway independently of the classical STING cascades [
56]. Upon cGAMP activation, STING was able to bind to and activate PERK, promoting PERK-mediated eIF2α phosphorylation [
56]. Through this pathway, DNA damage suppresses cap-dependent mRNA translation and turns cells to inflammation- and survival-biased translation programs, contributing to organ fibrosis and cellular senescence [
56].
Autophagy is a highly conserved intracellular degradation process used to remove damaged organelles, protein aggregates, and invading pathogens [
57]. Studies have shown that STING-mediated autophagy is required for innate immunity and the termination of cGAS-STING signaling [
50,
58]. Structurally, STING includes the light chain-3 (LC-3)-interacting regions (LIRs) through which STING can directly interact with LC-3 in order to regulate autophagy [
59]. Upon cGAMP-induced activation and dimerization, STING is translocated to ERGIC and binds to LC-3 and WD repeat domain, phosphoinositide interacting 2 (WIPI2) [
59,
60]. STING-containing ERGIC facilitates LC3 lipidation and the assembly of autophagosome depending on autophagy-related 5 (ATG5) and WIPI2 [
59,
61]. During this process, TBK1 and UNC-51-like kinase 1 (ULK1/ATG1), which were found to downregulate STING, are dispensable [
59,
61,
62].
4. Genotype
There is 81% structural similarity between hSTING and murine STING (mSTING) [
63], but hSTING has obvious heterogeneity. STING R232, not H232, is the most common hSTING allele, accounting for about 45% of the population [
64,
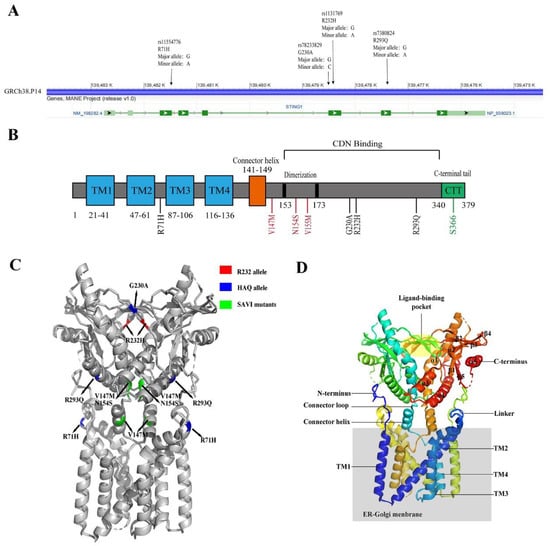
65]. Seema et al. found that R71H-G230A-R293Q (HAQ) was the second most-common hSTING allele (
Figure 2), accounting for about 16.07% of East Asians, about 7.78% of South Americans, and 6.75% of South Asians [
65]. R232/R232 is the predominant STING genotype of Europeans, and R232/HAQ is the most common hSTING genotype among East Asians [
65]. Approximately 10% of Europeans and 31% of East Asians have HAQ/HAQ, HAQ/H232, and H232/H232 genotypes [
65]. G230A and R232H are localized on the top of STING binding pocket, and R293Q is located at the bottom [
66]. R71H is located at a cytoplasmic loop facing the bottom, and SAVI mutants are located in the stem region of the binding pocket (
Figure 2) [
66].
Figure 2. The structure of hSTING. (A) The hSTING gene map (NCBI reference sequence: NC-000005.10), annotated with common SNPs. (B) Schematic diagram of the hSTING protein domain. Transmembrane domains are marked in blue, common human mutation points are marked in black, regions related to IRF3 activation are marked in green, and SAVI mutation points are marked in red. (C) The crystal structure of the hSTING protein (PDB: 7SII). R232 is marked in red, the HAQ mutation point is marked in blue, and the SAVI mutation point is marked in green. (D) Representation of the STING structure (PDB: 7SII). The secondary protein structure, N-terminus, and C-terminus are numbered. The ER–Golgi membrane is colored in gray.
A recent study suggested that HAQ and H232 might be STING loss-of-function alleles [
65]. Neither IRF3 nuclear translocation nor IFN-β production was observed in B cells that were homozygous H232 or HAQ/H232 stimulated with 2′,3′-cGAMP cyclic di-AMP (CDA), cyclic di-GMP (CDG), or RpRp-ssCDA [
65]. Although the activity of STING HAQ was greatly reduced, this did not translate into a loss of function [
67]. STING HAQ presented a more than 90% loss in its ability to stimulate IFN-β secretion due to R71H and R293Q in HAQ [
64]. The C292 and C88xxC91 near R293Q and R71H also played important roles in IFN-β stimulation [
64]. Compared with HAQ-expressing cells, the Q293-expressing cells displayed a mild deficiency of IFN-β stimulation [
64]. Furthermore, the H71 allele resulted in a more-severe deficiency in IFN-β stimulation than the Q293 allele [
64]. However, the H71 allele alone stimulated IFN-β secretion more effectively than HAQ [
64]. The defective IFN-β production capacity in HAQ carriers may be associated with the low level of STING protein expression and TMEM173 transcription. In addition, H232 has low binding affinity with CDNs [
65]. The STING expression in H232/H232 B cells is comparable to that in R232/R232 cells, while it is reduced in HAQ/H232 because of the HAQ allele [
65].
It has been reported that SAVI patients with HAQ had SAVI symptoms, but the onset of the disease was delayed (by approximately 3 years) [
68]. STING HAQ is associated with increased susceptibility to Legionnaires’ disease. Compared with the healthy people, the proportion of STING HAQ was raised in two groups of Legionnaires’ disease patients, while STING R232H was not increased [
67]. Although the replication of
Legionella pneumophila in wild-type (WT) STING and HAQ-type cells are similar, the production of IFN-β and IP-10 was reduced in homozygous HAQ cells, which were stimulated with cGAMP,
Legionella infection, synthetic DNA, and bacterial DNA, and this effect did not occur in response to the TLR7/8 agonist Resiquimod [
67]. Furthermore, the expression of IFN-β and IL-1β of heterozygous HAQ carriers was partially decreased [
67]. Therefore, the differences in STING expression and function found in individuals with HAQ allele may affect the therapeutic effect of STING-target treatment.
5. DNA Sensors Upstream of STING
Besides cGAS, DEAD-box RNA helicase 41 gene (DDX41) and IFI16 have been confirmed to participate in DNA recognition and get involved in STING activation [
69,
70,
71,
72].
The cGAS C-terminal domain includes a unique zinc band vital to binding to DNA [
73]. cGAS can bind to DNA pentose phosphate backbone and form a 2:2 cGAS-DNA complex in order to produce cGAMP and activate STING, thereby releasing IFN-β [
74]. There are effects of the type and length of nucleic acids bound to cGAS and their status on cGAS activity. DNA base oxidation induced by UV irradiation cannot affect the binding ability with cGAS [
75]. Single-stranded DNA (ssDNA) and double-stranded RNA (dsRNA) binding to cGAS cannot rearrange the structure of the catalytic pocket and thus cannot induce the production of cGAMP [
72,
75,
76]. The cGAS-STING pathway preferentially responds to long fragments of cytosolic DNA (>45 bp), which is related to cGAS K187 and L195 [
77]. Compared with murine cGAS, human cGAS K187 and L195 mutations result in increased sensitivity to long DNA fragments and decreased sensitivity to short DNA fragments [
77]. cGAS activity is also affected by post-translational modifications and the ionic environment. Mn
2+ can improve the sensitivity and activity of cGAS towards dsDNA while increasing the binding affinity of STING to cGAMP [
78]. Zn
2+ can improve recombinant cGAS activity in the buffer, which can be partially replaced by Mn
2+ and Co
2+ [
76].
IFI16 is a member of the PYRIN and HIN200 domain-containing (PYHIN) protein family [
79]. p204 is the mouse PYHIN most similar to IFI16, containing a pyrin domain and two DNA-binding HIN domains, which can directly bind to viral dsDNA to produce IFN-β [
71]. IFI16/p204 is considered to be the first PYHIN protein involved in IFN-β production and cellular senescence [
71,
80]. As the DNA-sensing protein, IFI16/p204 cooperates with cGAS in cGAMP-induced STING activation to defend from viral infection [
81,
82]. Under normal conditions, IFI16 is located in the cell nucleus [
83]. Upon the recognition of viral DNA, IFI16 polymerizes, forms an inflammasome complex, and is transported to the cytoplasm [
71,
84]. In the cytoplasm, IFI16 recruits TBK1 to STING and interacts directly with STING through the PYRIN domain, thereby initiating STING-IRF3 activation and promoting IFN-β production [
71,
82,
84]. In turn, STING and IFI16 can also regulate the one another’s protein levels in order to avoid continuous activation [
16,
17]. Furthermore, IFI16 can detect nuclear DNA damage and participate in the activation of the STING-NF-κB pathway with ATM independently of cGAS [
53].
DDX41, a helicase belonging to the DEXDc family, is the DNA sensor of myeloid dendritic cells (mDCs) [
69]. DNA can bind directly to the DDX41 DEAD domain, while the STING TM2, TM3, and TM4 domains can interact with DDX41 [
69]. Moreover, Walker A and B sequences of DEAD play indispensable roles in interacting with dsDNA or STING [
69,
84]. DDX41 can recognize dsDNA and interact with the STING-TBK1 complex to regulate IRF3 activation [
84]. Furthermore, DDX41 can interact with bacterial c-di-GMP and cGAMP to regulate the type I IFN signaling [
84,
85].
6. The Regulation of the STING Pathway
STING CTT has a self-inhibition effect on STING [
44]. The recruitment of TBK1 alone is insufficient to activate IRF3. STING CTT S366 is a vital site for IRF3 binding and activation [
36]. TBK1 can trigger the phosphorylation of STING CTT S366, providing a platform for IRF3 recruitment and TBK1 autophosphorylation. When S366 is mutated, IRF3 activation is blocked [
86]. Although STING CTT is indispensable for the STING-TBK1-IRF3 pathway, it is not vital for activating the STING-NF-κB pathway. The dmSTING (amino acids 147–343) of Drosophila lacks CTT but retains the ability to activate the NF-κB pathway through transcription factor Relish [
87].
Upon dsDNA stimulation, STING colocalizes with autophagy-related gene 9a (ATG9A) and LC-3 and conducts membrane trafficking in an ATG9A-dependent manner [
88]. ATG9A restricts innate immune responses by disrupting the binding between STING and TBK1 [
88].
After TBK1 activation, STING induces autophagy in order to degrade itself and p-TBK1 [
59]. At activation, STING can form a complex with adaptor protein complex 1 (AP-1) at the Golgi through the CTT domain, especially phosphorylated S366, controlling the degradation of STING through the endolysosomal system [
48]. STING S366 can also be phosphorylated by ULK1/ATG1, inducing STING degradation and preventing sustained activation by autophagy [
62]. Additionally, STING residues 330–334 play significant roles in autophagy induction. Mutations of L333A and R334A can inhibit LC-3 esterification, TBK1 and IRF3 phosphorylation, and cGAMP-induced STING and LC-3 puncta formation [
61]. Unc-93 homolog B1 (UNC93B1) is also able to interact with STING, reducing STING’s stability by delivering STING to the lysosomes for degradation [
89,
90].
Moreover, human epidermal growth factor receptor 2 (HER2) directly phosphorylates TBK1 by recruiting AKT serine/threonine kinase 1 (AKT1) and hinders the STING-TBK1 interaction and TBK1 K63-linked ubiquitination, attenuating the STING signaling pathway [
91,
92]. Likewise, when STING C148 was mutated, the affinity of STING to cGAMP was decreased, and the downstream pathway was inhibited [
44].
Within the ubiquitin–proteasome pathway, host cells can degrade STING-pathway inhibitor factors in order to activate the pathway while limiting the continuous activation of the pathway so as to avoid self-injury caused by excessive inflammatory responses (
Figure 3) [
93]. Autocrine motility factor receptor (AMFR) and insulin-induced genes 1 (INSIG1) are ER-protein and can form a complex which functions as E3 ubiquitin ligase [
94]. An AMFR and INSIG1 complex has been reported to catalyze STING K27-linked polyubiquitination [
94]. Upon this modification, TBK1 is recruited, inducing STING transfer to the perinuclear body and enhancing STING pathway activation [
94]. Contradictorily, ER-localized E3 ubiquitin ring finger protein 5 (RNF5) and tripartite motif protein family member 29 (TRIM29) are able to catalyze the K48-linked ubiquitination of STING K150 and K370, respectively [
33,
95], resulting in the degradation of STING. Moreover, cylindromatosis (CYLD), a deubiquitinating enzyme, can specifically remove K48-linked ubiquitination, stabilizing the STING protein [
96]. Tetsuo et al. found that STING K150 was not only the site of the K48-linked ubiquitination of STING but also an essential amino acid residue for K63-linked ubiquitination [
97]. TRIM56 catalyzes K63-linked ubiquitination of Lys150, while TRIM32 catalyzes K63-linked ubiquitination of K20/150/224/236, respectively, promoting STING dimerization and interaction with TBK1 [
97,
98]. Additionally, it was found that blocking post-translational modifications of STING [
99], like ubiquitination at K224 and K228, only inhibited the STING-IRF3 pathway without affecting NF-κB-related responses [
100,
101].
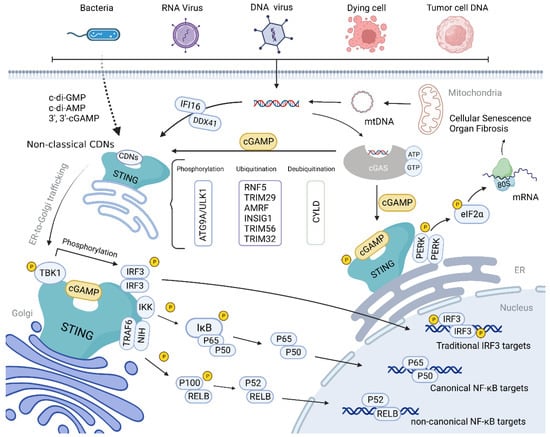
Figure 3. Schematic diagram of the cGAS-STING signaling pathway. cGAS can recognize abnormally exposed cytoplasmic DNA molecules, including viral and bacterial DNA, DNA produced by the reverse transcription of RNA viruses, and DNA produced by self-cell damage. It can catalyze the synthesis of 2′, 3′-cGAMP, which specifically binds to STING dimer for oligomerization. After activation, STING is translocated to the Golgi via ERGIC, during which TBK1 and IRF3 are recruited, and this complex induces an immune response by phosphorylating IRF3 or NF-κB. In addition, STING can activate PERK and promote the phosphorylation of eIF2α, inducing translation program transformation. Autophagy, ubiquitination, recruitment inhibition, mutation, and other pathways can affect the STING pathway.
7. STING Inhibitors
The over-release of type I IFN is a significant trigger of numerous IFN-related diseases, including SLE, AGS, and SAVI. Down-regulating the over-activated STING pathway can reduce the type I IFN release, help the immune system return to normal, and maintain the body’s dynamic balance. However, research on STING inhibitors is in the early stages, and no compounds have yet entered clinical trials (Table 1).
Table 1. An overview of STING inhibitors.
|
Inhibitor
[Ref]
|
Binding Sites
|
Molecular
Mechanism
|
Biological Activity
|
|
Nitro-fatty acid
Derivatives
[102]
|
C88, C91 at palmitoylation site and H16 in N-terminus
|
Covalently bind to the STING cysteines residues, block STING palmitoylation and inhibit STING activation
|
N.D.
|
|
C-176/178/170 and
H-151
[103]
|
C91 at palmitoylation site
|
IC50 (H-151) = 134.4 nM
(HFFs cells)
|
|
BPK-21/25
[104]
|
C91 at palmitoylation site
|
ISRE-Luc activity (BPK-25) = 3.2 μM (THP1 cells)
|
|
Astin C
[21]
|
CDN binding site
|
Compete with cGAMP for the CDNs binding pocket and inhibit STING activation
|
IC50 = 3.42 ± 0.13 μM (MEFs cells)
|
|
Compound 18
[105]
|
CDN binding site
|
IC50 = 68 nM
(STINGHAQ);
IC50 = 11 μM
(THP1 cells)
|
|
SN-011
[106]
|
CDN binding site
|
IC50 = 502.8 nM (HFFs cells)
|
|
Palbociclib
[107]
|
CDN binding site
|
Interact with STING CTD and block STING dimerization
|
IC50 = 0.81 ± 0.93 μM (HEK293 cells)
|
|
Compound 30
[108]
|
CDN binding site
|
Undetermined
|
IC50 = 1.15 μM (RAW264.7 cells)
|
|
6,5-heterocyclic derivatives
|
Unknown
|
Undetermined
|
IC50 ranges from 30 μM to less than 10 nM
|
|
SP23
[109]
|
Palmitoylation site
|
Promote the degradation of STING via ubiquitin-proteasome pathway
|
DC50 = 3.2 μM (THP1 cells)
|