Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
疱疹病毒是感染人类和动物的主要病原体。操纵大基因组对于探索特定基因的功能和研究疱疹病毒的发病机制以及开发新型抗病毒疫苗和治疗方法至关重要。细菌人工染色体(BAC)技术显着提高了疱疹病毒研究人员操纵病毒基因组的能力。
- herpesvirus
- bacterial artificial chromosomes
- gene editing
- virus mutant selection
1. 简介
基因组操作是研究病毒基因功能的有效方法,可以帮助科学家了解病毒的生物学,如发现毒力因子,探索开发预防和治疗病毒感染的新型疫苗和抗病毒药物的新靶点。疱疹病毒是一组双链DNA(dsDNA)病毒,可引起终身持续感染,是人类和各种动物的主要病原体。疱疹病毒的基因组长度在125至295千碱基之间[1],这使得基因组操作变得困难。
在过去的几十年里,科学家们开发了几种操纵疱疹病毒基因组的方法,如λ-red、Cre-loxp、CRISPR-Cas9等,有助于了解疱疹病毒的发病机制,开发新型疫苗和抗病毒药物[2,3]。传统上,将含有选择盒和侧翼同源区域的线性DNA片段或环状质粒转移到疱疹病毒感染的细胞中,通过重组编辑疱疹病毒的基因组,然后使用不同的标记(不同的选择盒)选择重组病毒[4,5,6,7].这种方法效率低。此外,它对后代病毒产生的依赖导致可以编辑的基因范围有限。此外,用于识别功能性重组病毒的繁琐筛选过程限制了其应用[3]。因此,必须探索能够提高效率和扩大可编辑基因范围的替代技术。基于大肠杆菌因子F构建的细菌人工染色体(BAC)为编辑包括疱疹病毒在内的大基因组提供了重要的技术平台[8,9,10,11]。基于BAC的基因编辑策略独立于后代的产生,筛选效率高于常规策略[12,13,14]。首先,BACs可以容纳100-300 kb外源基因片段的插入和稳定遗传。F因子有助于维持大肠杆菌中的低拷贝数(每个细胞1-2个拷贝),从而大大降低了筛选的难度。同时,各种基因编辑技术在大肠杆菌中的开发,与传统方法相比,大大提高了疱疹病毒BACs的编辑效率。编辑过的BAC可以重新引入真核细胞以重建疱疹病毒突变体,以进一步研究它们在细胞中的生物学行为。此外,重组工程病毒的选择不依赖于后代病毒的产生,允许编辑长期维持潜伏感染的γ疱疹病毒基因组[13,15]以及操纵产生后代病毒所必需的基因[11]。
2. 基于BAC的疱疹病毒基因组编辑技术
2.1. RecA 重组技术
RecA重组技术是由RecA重组酶和相关辅助蛋白(如RecBCD、RecFOR、RuvABC等)组成的细菌内源同源重组系统。RecBCD是一种多功能酶复合物,由RecB,RecC和RecD组成。当与双链供体DNA上的切口结合时,RecB和RecD分别表现出3′至5′和5′至3′解旋酶活性,它们共同导致互补双链DNA的开放(图1a)[16]。然后,RecC可以介导序列中气位点的识别,从而引起变构改变,导致运动速度降低(图1b)[17]。此时,RecB还具有核酸外切酶活性,其介导3′末端单链DNA的生成(图1c)[18,19,20]。RecA重组酶与核酸外切酶产生的单链DNA位点结合,并参与鉴定BAC上同源区域的过程,以便随后退火和相互作用。成功相互作用后,核蛋白丝侵入dsDNA并进行链交换以形成异质双链DNA(图1d)。最后,RuvABC有助于催化分支迁移和异源双链DNA的降解,导致同源重组(图1e)[21]。
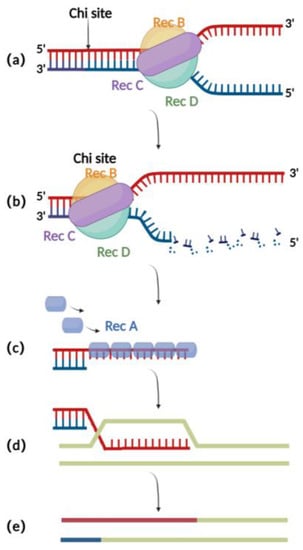
图1.RecA重组技术示意图:(a)RecBCD与双链DNA末端结合并促进解开。(b)在气位点,在3'末端产生单链DNA。(c)RecA蛋白在3'末端与单链DNA结合。(d)ssDNA-RecA侵入完整的同源双链DNA进行链交换。(e) 重组完成。
尽管RecA介导的同源重组方法显示出更高的效率(10−6到 10−4)与编辑真核细胞中疱疹病毒基因组的传统方法(表1)相比[22],它仍然具有明显的缺点。一个主要问题是,由于疱疹病毒基因组中存在重复序列,RecA的表达可导致疱疹病毒BAC克隆不稳定,进而导致病毒基因组突变(表1)[1]。此外,RecA介导的重组通常使用500 bp至3 kb长的同源臂(表1)[7,23,24],导致构建重组所需的穿梭质粒的过程很麻烦。这些缺点限制了RecA介导的重组的应用。
表 1.基于BAC的疱疹病毒基因组编辑技术的优缺点。
| 编辑技巧 | 同源臂的长度 | 编辑效率 | 编辑网站的要求 | BAC的稳定性 |
|---|---|---|---|---|
| RecA 重组 | 500 基点–3 千基点 | 10−6到 10−4 | 没有 | 导致 BAC 突变 |
| λ-红色复合 | 30–50 基点 | <1% | 没有 | 保持了BAC的稳定性 |
| 基本编辑 | 不需要 | 接近 100% | PAM 网站和基本编辑网站;只能进行基本编辑 | 保持了BAC的稳定性 |
2.2. λ-红色复合技术
由λ-red重组系统介导的同源重组是目前编辑疱疹病毒BAC克隆最广泛使用的重组技术[2,25]。λ-红色重组系统来源于噬菌体λ,由Gam、Exo和Beta蛋白组成。Gam蛋白是Exo蛋白和β蛋白的辅助蛋白。Gam蛋白可以抑制Rec BCD与dsDNA末端的结合,从而抑制Rec BCD外切酶的功能,防止外源性双链DNA的降解(图2a)[26,27]。Exo蛋白可以结合到dsDNA供体的末端,dsDNA供体在靶基因的两侧都包含同源片段。同时,Exo蛋白具有5′至3′核酸外切酶活性,在3′末端产生一段单链DNA(图2b)[28]。β蛋白在λ-红同源重组过程中起决定性作用。作为单链DNA结合蛋白,β蛋白与Exo蛋白产生的单链DNA结合。β蛋白的结合增强了供体DNA片段的退火和复制疱疹病毒BAC靶位点的同源序列(图2c)。DNA的复制完成了同源重组(图2d)[29,30]。
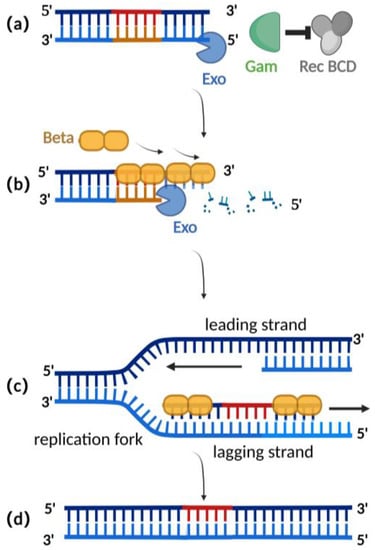
Figure 2. Schematic diagram of λ-red recombination technique: (a) Gam protein inhibits the activity of Rec BCD exonuclease; (b) Exo protein creates a stretch of single-stranded DNA at the 3′ end; (c) Beta proteins bind here, facilitating annealing interactions between the donor DNA fragment and the homologous sequence of the target site; (d) homologous recombination is complete with the replication of DNA.
Compared with the RecA recombination technique, the λ-red recombination technique avoids the risk of a partial deletion of the herpes virus genome in BAC during the recombination process as only homologous double-strand ends can be used as a substrate (Table 1). Additionally, this method typically only requires 30–50 base pairs of homologous arms for recombination (Table 1), making it easier to obtain the donor through techniques such as oligonucleotide synthesis or polymerase chain reaction (PCR), eliminating the need for shuttle plasmids as required in RecA recombination [31,32]. Importantly, the recombination efficiency mediated by the λ-red recombination technique has been improved to a maximum of 0.68% when the donor is double-stranded DNA (Table 1) [33,34,35].
2.3. Base Editing Technique
CRISPR/Cas 9 is a powerful genome editing technique [36] that has been successfully applied in a wide range of eukaryotic cells, including human cell lines, embryonic stem cells, mice, Arabidopsis, and Drosophila [37,38,39,40]. In 2013, Jiang et al. successfully edited Streptococcus pneumonia’s genome using CRISPR/Cas-9-only gene editing [41]. Since then, it has been successfully employed in a variety of prokaryotic species as well [42,43]. The versatility and effectiveness of CRISPR/Cas9 in modifying the genome make it a valuable tool for a wide range of scientific and medical applications.
Due to the lack of the non-homologous end-joining (NHEJ) pathway [44,45,46,47] and the low efficiency of their endogenous homologous recombination system, it is difficult to achieve stable genome editing in most bacteria using CRISPR/Cas9 gene editing technology alone [41,48,49,50]. Until now, there was still no publication reporting CRISPR/Cas 9-based gene editing technique in the stably preserved herpesvirus BAC gene in E. coli. This highlights the need to explore alternative approaches to achieve efficient and reliable gene editing in these organisms.
Recently, scientists have constructed an efficient gene editing method by combing CRISPR/Cas 9 gene editing technique with precise base editing technology. This method consists of an sgRNA and a complex that includes modified Cas9 proteins, cytosine deaminases, and an uracil glycosylase inhibitor (UGI) [51]. Unlike wild-type Cas9 proteins, the modified Cas9 is catalytically dead, lacking endonuclease activity. Therefore, it can only facilitate genome targeting via sgRNA but cannot induce a double strand break (DSB) due to the absence of cleavage activity (Figure 3a). Cytosine deaminase converts the specified cytosine (C) site to uracil (U) (Figure 3b). At this point, uracil glycosylase inhibitors can prevent the excision of intermediate product U, increasing the efficiency of converting C to T on the DNA chain, ultimately achieving single-base precise editing of C to T and G to A (Figure 3c).
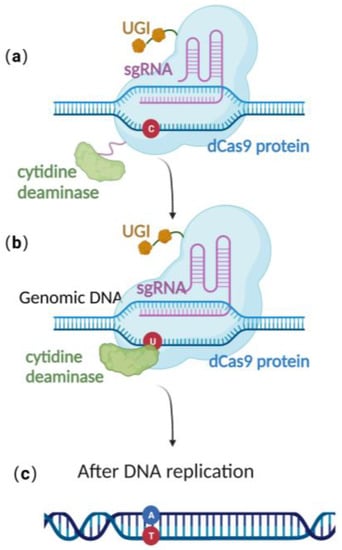
Figure 3. Schematic diagram of base editing method: (a) dCas9 mediates targeting without DSB formation; (b) cytosine deaminase converts C to U; (c) single-base precise editing is complete with the replication of DNA.
Base editing allows for site-directed mutagenesis of multiple prokaryotic genomes [52], including E. coli [22] and even herpesvirus genome BACs preserved in E. coli [53]. Zheng et al. [22,53] utilized this technology, directly converting cytidine (C) to uridine (U) at specific positions on the US8 and UL34 genes of the pseudorabies virus genome BAC, thus achieving the premature termination of the corresponding genes and approaching 100% editing efficiency (Table 1). Due to the modifications that occur in the base-editing window, many studies have demonstrated that base editing is associated with off-target effects [54,55], thereby limiting its practical application. To effectively utilize base editing, optimization of the cytidine deaminase and/or UGI is necessary [56,57]. Despite this, the technology has the advantage of directly editing the target site nucleotide without the need for donors. It provides an efficient alternative method for point mutation editing.
Moreover, the novel combined editing technology that has emerged in bacterial genome editing but has not yet been applied to BACs and may also provide insights for future BAC editing. Combining CRISPR/Cas 9 with λ-red recombination could increase the efficiency of recombination in editing the genome of E. coli, with reported knockout efficiency of up to 100% for deletion lengths of up to 3.4 kb, which is higher than in previous reports [48,58,59]. The use of this system could potentially offer a new approach for efficient editing of the BAC of herpesvirus genomes that are stably stored in E. coli.
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms11030589
This entry is offline, you can click here to edit this entry!