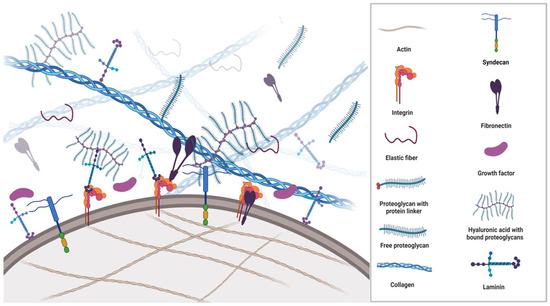
All cells synthesise, secrete, and degrade the extracellular matrix (ECM) occupying the space between them. Apart from being passive mechanical support for cells, the ECM is an extraordinarily complex and highly dynamic macromolecular meshwork of proteins, glycoproteins, proteoglycans, water, minerals, and a multitude of bioactive molecules that determine the phenotypes and molecular functions of the cells it surrounds (Figure 1). The interaction between ECM components, ECM-bound factors and cell surface receptors plays a crucial role in mediating cell adhesion and signalling that regulates multiple biological processes. Additionally, the ECM caters to the three-dimensional architectural structures of organs.
2. Components of ECM
ECM is a composite of cell-secreted macromolecules that include fibrous proteins providing the tissues tensile strength and glycoproteins and proteoglycans providing resistance to compression and deformation. Importantly, these molecules participate in multiple signalling pathways and are described in the subsequent section.
3. Fibrous ECM Proteins
3.1. Collagen
Collagen is a polypeptide structure produced by fibroblasts. Except for the brain, collagen is the most abundant protein throughout the human body and the most significant protein in the ECM [
11]. Collagen is the major component of most connective tissues supporting and contributing to the three-dimensional from of organs. In addition, collagen plays an important role in various physiologic processes that include angiogenesis, haemostasis, and mineralisation, as well as in common pathologies such as cancer, fibrosis, and cardiovascular diseases [
12].
In solid cancers, collagen deposition not only creates a barrier for cytotoxic immune cells and increases therapy resistance but also provides a rich source of exploitable metabolic fuels for cancer cells [
13].
Following synthesis and assembly in the endoplasmic reticulum, the precursor peptide procollagen is packaged and exocytosed into the extracellular space. In the extracellular space, propeptide domains at the carboxy- and amino terminals of the procollagen are cleaved off by MMPs to modify the fibril shape and prevent lateral growth. The formation of the mature collagen microfibril requires binding to the N-terminus of fibronectin [
14].
Each collagen fibre is made up of several subtypes. Defined by their bonds and amino acid repeats, twenty-eight different types of collagen composed of at least 46 distinct α-chains have been identified in humans [
15]. Nearly 50% of amino acids incorporated into collagens are proline and glycine, which have important roles in the regulation of energy production, protein synthesis, redox balance, and intracellular signalling [
16].
Collagen can be divided into fibrillar collagens type 1, 2, 3, 5, 11, 24, and 27 and non-fibrillar collagen type 4 (basement membrane); 6 (beaded filaments); 7 (anchor fibres); 8, 10 (short chain); 9, 12, and 14 (fibril-associated collagens with interrupted helices or FACIT); and type 13 (transmembrane collagen). The most abundant is collagen type 1, found in the skin, bones, and tendons [
17].
Mutations in collagens 1, 2, 3, 9, 10, and 11 result in a broad range of ailments affecting cartilage, bones, and blood vessels, including osteogenesis imperfecta, various types of chondrodysplasia, Ehlers–Danlos syndrome types 4 and 7, and some cases of osteoarthritis, osteoporosis, and familial aneurysms [
18].
3.2. Elastin
Elastin is a fibrillar hydrophobic matrix protein that, in contrast to collagen, is able to stretch eight times its resting length [
19]. Elastin provides flexibility to blood vessels, skin, lungs, and ligaments. It is synthesised by fibroblasts, vascular smooth muscle cells [
20], smooth muscle cells, and several types of epithelial cells. Being the primary ECM protein in arteries, where it amounts to ~50% of the weight [
21], it has an impressive ability to withstand the mechanical stress of more than 3 billion expansions and contractions during an 80-year life cycle.
The precursor protein, tropoelastin, is secreted with a chaperone molecule that facilitates the correct folding of the protein before it is incorporated into the highly flexible elastin strands. Similar to other ECM proteins, such as collagens, mature elastin is extensively cross-linked with other elastin molecules to form sheets and fibres [
22]. Elastic fibres are composed of approximately 90% elastin, whilst the remaining components are primarily comprised of fibrillin glycoproteins. Due to its unique structure, extensive cross-linking and durability, elastin does not undergo significant turnover in healthy tissues where it has a half-life of more than 70 years [
23]. It is primarily deposited during prenatal development and childhood and is rarely synthesised during adulthood [
24]. Aberrant expression of elastases and degradation of elastin trigger the release of elastokines (fragments of matrix proteins with cytokine-like properties) that promote angiogenesis and regulate cell adhesion, chemotaxis, migration, and proliferation. Much of the elastokine effects are mediated by membrane elastin receptor complexes [
23] that trigger signalling pathways involving the extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) and serine/threonine-protein kinase (AKT) activation [
25]. Elastases belong to the enzyme classes of MMP, aspartic proteases, serine proteases, and cysteine proteases. The destruction of elastin promotes the development and progression of different pathological conditions, including chronic obstructive pulmonary disease, atherosclerosis, vascular aneurysms, and cancer. In lung and colon cancer, the degradation of the matrix and fragmentation of elastin was found mainly to occur at the invasive front, and the expression levels of MMP also correlated to the metastatic potential of these cancers [
26,
27].
4. The Glycoproteins
Glycomics is an important new frontier in life science research. Similar to proteoglycans, glycoproteins are composed of proteins with attached saccharide chains; however, the glycoprotein side chains are much shorter than the saccharide chains in proteoglycans. They contain no (or few) repeating units and are usually branched. The two most important ECM glycoproteins are fibronectin and laminin.
Glycoproteins often act as connecting molecules that bind other ECM molecules, GF, and receptors. They have N-linked and O-linked saccharide sidechains, with the N-linked chains being connected to -NH2 on asparagine residues in the protein and O-linked chains to the -OH on the serine/threonine residues. N-linked and O-linked glycoproteins are mainly located on the cell membrane, where they play crucial roles in the cell–cell communication, adhesion, migration, proliferation and healing processes; they may also exist as secreted proteins.
4.1. Fibronectin
Within the body, fibronectin exists as soluble plasma glycoproteins (synthesised by hepatocytes and secreted into the blood) and as insoluble cellular fibronectin (a fibrillar cross-linked structure on the cell membranes). It is responsible for cell adhesion, proliferation, migration, and the deposition of ECM proteins [
28].
The basic structural unit of fibronectin is a dimer composed of two nearly identical polypeptide chains linked by a pair of disulphide bonds. Fibronectin fibrils serve as mechanical links between the cytoskeleton and the surrounding ECM. It primarily binds to actin-anchored integrins on the cell membrane (
Figure 1). Mediating the adhesion of BM components to ECM structures, integrins are heterodimeric (α and β subunits) cell-surface receptors and bi-directional transducers of biochemical signals and mechanical forces acting on the ECM. The α and β subunits both have a cytoplasmic tail, a transmembrane domain, and a large extracellular domain that bind numerous ECM ligands. The anchorage to the ECM is required for normal cells to enter the S phase, even in the presence of GF. If cells detach from their integrin ligation points and lose the sense of their mechanical environment, they undergo a specific type of apoptosis, anoikis (Greek for homeless). Resistance to anoikis is a characteristic feature of tumour cells that enables them to survive under non-adherent conditions [
29,
30,
31].
The connection between the ECM and cytoskeleton stimulates cell proliferation and angiogenesis through pathways that include ERK 1/2 phosphorylation, dysregulation of the HIPPO (tumour suppressor) pathway, and suppression of apoptosis through the nuclear factor kappa B (NF-κB) or the phosphoinositide 3-kinase (PI3kinase)/AKT pathway [
32].
Fibronectin fibrillogenesis is initiated by cytoskeleton-derived tensional forces transmitted across transmembrane integrins, typically α5β1 [
33]. During this process, soluble molecular fibronectin is irreversibly assembled into insoluble fibrils that stretch up to four times their resting length, which implies domain unfolding and subsequent ECM remodelling [
34]. Fibronectin fibres are proposed to be held together by hydrogen and disulphide bonds; however, catalytic agents such as thermolysin, plasmin, thrombin, trypsin, cathepsin D, and chymotrypsin can cleave them.
Fibronectin fibrillogenesis and collagen fibrillogenesis have a complex relationship, with fibronectin regulating the assembly of collagen and vice versa [
35]. How the production, organisation and matrix deposition of fibronectin are regulated by tumour cells is less understood as the turnover of fibronectin is largely unexplored [
36].
Interacting with other ECM proteins, including GF, glycosaminoglycans, cell surface receptors and other fibronectin structures, fibronectin provides key mechanical and chemical signals to induce differentiation and epithelial-mesenchymal transition (EMT) [
37].
Transforming growth factor β (TGFβ), fibroblast growth factor (FGR), platelet-derived growth factor (PDGF), hepatocyte growth factor (HGF), and vascular endothelial growth factor (VEGF) have multiple binding sites within fibronectin. The binding of TGFβ1 to fibronectin fibrils was shown to upregulate EMT [
37], whereas dysregulation of fibronectin promoted tumorigenesis and fibrosis, with the expression levels of fibronectin being significant prognostic factors in several cancers [
38,
39].
Hypoxia-induced factors upregulated in tumour cells stimulate endogenous FN synthesis. Intercellular signalling between tumour cells and protumorigenic stromal cells, such as tumour-associated macrophages, cancer-associated fibroblasts, and myeloid-derived suppressor cells drive persistent FN deposition and remodelling of the ECM that facilitate growth and dissemination [
40,
41,
42].
4.2. Laminin
Laminins are one of the major glycoproteins in the basement membranes that glue cells and tissues together and regulate cellular activities and signalling pathways. Structurally, laminins are cross-shaped, trimeric glycoproteins of 400–800 kDa in size and composed of a few distinct domains, of which 16 different combinations have been identified. Primarily involved in tissue repair and wound healing [
43,
44,
45], all laminin complexes have a high affinity for GF through their heparin-binding domains; thus, apart from contributing to the anchoring of cells, laminin is a storage facility for GF whose release determines cell differentiation, survival, shape, and motility [
46]. In hepatocellular carcinoma, laminin was found to be involved in EMT and disease progression [
47]. The association between ECM proteins and GF is shown in
Table 1.
Table 1. Growth factors and their association with ECM proteins.
5. Proteoglycans
The proteoglycans (mucoproteins) are composed of a protein core covalently attached to glucosaminoglycans (mucopolysaccharides), such as chondroitin sulphate, heparan sulphate or keratan sulphate (
Figure 1). Proteoglycans have excellent water retention, gel-forming and space-filling functions [
48], conveying resistance to compression and deformation to cells. Although one of the least abundant components in the ECM, they are integral in maintaining a healthy ECM.
Syndecans are transmembrane proteoglycans with heparan and chondroitin sulphate chains attached to their extracellular domain. They may also exist as soluble extracellular domains. Similar to many proteoglycans, they interact with a multitude of ligands, such as GF, adhesion receptors, proteinases, cytokines, chemokines and other ECM proteins to initiate downstream signalling responsible for proliferation, adhesion, angiogenesis, and inflammation [
49]. Elevated levels of syndecan expressions in cancer can be correlated with poor outcomes, e.g., of Syndecan-1 in breast cancer and of Syndecan-2 in colorectal cancer, where it is highly associated with metastasis [
50].
Hyaluronic Acid
Hyaluronic acid (HA or hyaluronan) is a hydrophilic glycosaminoglycan. HA synthases in the cell membrane mediate the alternate addition of glucuronic acid and N-acetylglucosamine in a growing chain of thousands of disaccharides [
51] that are translocated out of the cell during biosynthesis. HA is abundantly present in the ECM of weight-bearing joints and the interstitial gel [
52,
53].
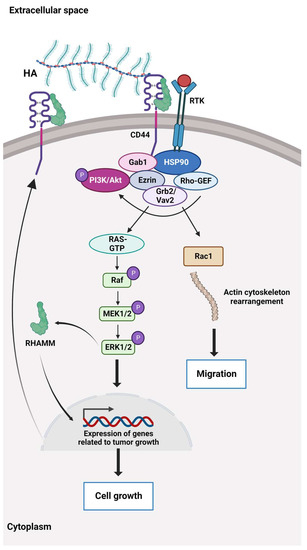
HA binds to the cluster of differentiation 44 protein (CD44), a transmembrane receptor that participates in many physiological and pathological processes by interacting and activating key signalling cascades (
Figure 2). Ligation of CD44 initiates the expression of genes related to tumour growth, proliferation, and survival, and its ligation with HA induces cytoskeletal rearrangements and membrane ruffling that leads to active cell migration [
54,
55]. Further, CD44 serves as a marker for several types of stem cells [
56,
57].
Figure 2. The HA-dependent CD44 signalling. CD44 and the receptor for hyaluronic acid (HA)-mediated motility (RHAMM) are the main HA receptors. They are commonly overexpressed in cancer, where they activate signalling pathways related to disease progression. CD44 also interacts with other ligands, such as collagens and matrix metalloproteinases, and as such, a multifunctional receptor mainly involved in proliferation, differentiation, migration, and angiogenesis. In contrast to membrane-bound receptors containing signalling domains, RHAMM (CD168) does not contain signal sequences. RHAMM is localised inside the cell and is exported to the cell surface in response to stimuli such as cytokines, including TGF-β. Extracellularly, RHAMM associates with CD44, and when HA binds to these cell surface receptors, it triggers several signalling pathways and complex formation between CD44 and its co-receptors and increases the expression of TGF-β receptors. Activation of downstream effectors, e.g., Akt, PI3K, MEK1/2, ERK1/2, and Ras/Raf/Rac, results in the expression of a variety of inflammatory cytokines and activation of a feedback loop augmenting cell surface expression of CD44/RHAMM. The CD44/RHAMM complex stimulates cell motility and increases angiogenesis by promoting the migration of endothelial cells towards the tumour. Further, these signalling events drive proliferation, invasion, and cytoskeletal rearrangements, leading to normal cell functions, such as fibroblast migration and immune cell function, and tumour growth and progression. ERK1/2: Extracellular signal-regulated protein kinases 1 and 2, Ezrin: kinase substrate protein, Gab1: PAR-1 kinase, Grb2/Vav2: growth factor receptor-bound protein2/guanine nucleotide exchange factor, GTP: Guanosine triphosphate, HSP90: Heat shock protein 90, MEK1/2: Mitogen-activated protein kinases 1 and 2, PI3K/Akt: Phosphoinositide 3 kinase/protein kinase B (originally Ak strain transforming kinase), Ras/Raf/Rac: Rat sarcoma virus protein/rapidly accelerated fibrosarcoma protein kinase/Ras-related C3 botulinum toxin substrate 1, Rho-GEF: Rho-guanine nucleotide exchange factors, RTK: Receptor tyrosine kinase.
HA turnover occurs in various molecular weights of HA distinguished by their polymer length, e.g., high-molecular-weight HA (Mw > 1.8 × 10
6 Dalton) and low-molecular-weight HA, Mw 4–10 × 10
5 Dalton), The molecular weight of HA determines its activities. High-molecular weight HA inhibits mitogenic processes and possesses anti-inflammatory effects, whereas low-molecular weight-HA shows protumorigenic effects by enhancing proliferation and inflammation [
58].
Within a solid tumour, HA is deposited by cancer-associated fibroblasts (CAF) and cancer cells and is a major structural component of the TME [
59].
HA plays a central role in cancer cell proliferation and migration. HA recruits tumour-associated fibroblasts, macrophages, and HA fragments to promote angiogenesis and immunosuppression, either directly or through macrophage protumorigenic polarisation [
60,
61]. Moreover, increased HA levels in the TME are associated with poor prognosis and survival in several cancers [
54,
62]. The recruitment and polarisation of macrophages may be used in future targeted anticancer therapies. Chimeric antigen receptors (CARs) have become a promising approach to increasing tumour cell recognition by cytotoxic immune cells. CAR-T cells are already in clinical use. In vitro, tumour-associated macrophages engineered with CAR constructs can be directed at tumour antigens and kill tumour cells by phagocytosis. Furthermore, the CAR-HER2-CD147 construct activates the expression of matrix metalloproteinases, degrades the ECM, and overcomes the physical barrier that prevents the infiltration of cytotoxic immune cells. As the degradation of the ECM may also support the dissemination of tumour cells, any treatment involving metalloproteinases must be calibrated meticulously [
63].
Second-generation CAR-M cells are in development. In addition to maintaining the characteristics of first-generation CAR-M technology, the goals of second-generation therapies comprise improving tumour-associated antigen presentation and T-cell activation [
64].
6. Sensing and Communication in ECM
Mechanotransduction allows living organisms to receive and respond to mechanical forces from the internal and external environment. Mechanically activated ion channels represent the primary mechanism for mechanotransduction that effectively converts mechanical stimuli into electrochemical signals. In the cell membrane, the transmembrane and mechanotransducing Piezo proteins (1 and 2) form ion channels that are activated by pressure. When pressure is applied to the cell membrane, the large, three-bladed propeller-shaped molecular complexes flatten and stretch, whereby the ion channels open and allow a flow of calcium and other positively charged ions through the central pore module into the cell, whereby biochemical signals are created.
Cells probe their environment mechanically via lamellipodia (membrane protrusions composed of a dense and dynamic network of actin filaments). Lamellipodia decode the mechanical feedback and resistance from their surroundings [
65] through membrane integrins and syndecans. These molecular complexes trigger intracellular signalling cascades involving the unfolding of proteins associated with the contractile actin cytoskeleton, such as Rho-associated protein kinase [
66]. When the actin protein polymerises to form filaments, it enables the cell to control shape and mechanics—as used by crawling and phagocytising immune cells and migratory tumour cells.
In cancer, the cytoskeleton signalling reduces the number of intercellular adhesion molecules, induces EMT, upregulates membrane integrins, and enhances the migratory potential [
7] and therapeutic resistance [
67]. The mechanotransduction that arises from changes in ECM stiffness shifts cytoskeletal dynamics and releases mechanosensitive molecules such as vinculins, paxicillins, and talins that also regulate adhesion and migration [
68,
69]. Mechanosensitive cytoplasmic proteins connect integrin complexes to the nucleus through linker protein complexes that allow a direct transmission from ECM to the nucleus as reviewed in [
70].
The mechanisms leading to gene transcription are not mapped fully. Yes-associated protein 1 (YAP) and WW-domain-containing transcription regulator 1 (TAZ) are transcriptional coactivators that are translocated to the nucleus together with β-catenin upon cytoskeletal tension [
71,
72,
73]. YAP/TAZ activation upregulates the genes associated with proliferation and dedifferentiation, and the nuclear translocation of β-catenin directly destabilises intercellular adhesions, all contributing to EMT for migration through the ECM [
74,
75]. Simultaneously, the levels of several integrins are upregulated in several tumours. Many signalling pathways are affected by ECM dysregulation, as shown in
Table 2.
Table 2. A non-exhaustive table summarising the expression levels of some of the main signalling pathways in various solid cancers that are affected by ECM dysregulation, such as aberrant collagen deposition, increased HA expression, and abnormal expression of laminin and fibronectin. It is important to note that the expression levels of signalling pathways may vary. They are not mutually exclusive, and as shown, they often overlap and interact with each other. Signalling pathways are also affected by the accumulation of genetic mutations and disease progression and may change their oncogenic drive. With progression, changes in the composition and organisation of the ECM may affect additional pathways. For example, in early-stage breast cancer, changes in collagen I and IV deposition can activate the PI3K/AKT and MAPK/ERK pathways, whereas in advanced stages, the deposition of different types of collagens, such as collagen VI, can activate the TGF-β pathway. Similarly, in lung cancer, the expression of hyaluronan is increased in advanced stages, leading to dysregulation of the Hippo pathway, which promotes tumour growth and metastasis. CRC: colorectal cancer, EMT: epithelial-mesenchymal transition, EGFR: epidermal growth factor receptor, ERK: extracellular signal-regulated protein kinase, FAK: focal adhesion kinase, HA: hyaluronic acid, HCC: hepatocellular carcinoma, MAP/ERK: microtubule-associated protein kinase; MAPK: mitogen-activated protein kinase, NSCLC: non-small cell lung cancer, PDAC: pancreatic ductal adenocarcinoma, PI3K/Akt: phosphoinositide 3 kinase/protein kinase B (originally Ak strain transforming kinase), Ras/Raf: rat sarcoma virus protein/rapidly accelerated fibrosarcoma protein kinase, RhoA/ROCK: Rho GTPase A/serine-threonine protein kinase, SMAD: Suppressor of Mothers against Decapentaplegic, Src: non-receptor cytoplasmic tyrosine kinase, YAP: Yes-associated protein, TGF-β: transforming growth factor. Wnt: wingless-related integration site.
Integrins activate various intracellular signalling pathways that promote cell survival, growth, and proliferation and targeting these molecules could be an effective strategy to strike at tumour cells. Integrins play key roles in various other diseases, such as ulcerative colitis, cardiovascular diseases and osteoporosis, but they have not been targeted extensively (reviewed in [
76]). Kindlin-2 is a widely expressed protein that is critical for integrin-mediated cell–ECM adhesion and signalling. Kindlin-2 localises to the adhesion sites where the ECM molecules are connected to the actin cytoskeleton and increase proline synthesis through interaction with pyrroline-5-carboxylate reductase 1 (PYCR1). PYCR1 is a key mitochondrial enzyme that facilitates the last step in glutamine-to-proline conversion, and its overexpression of PYCR1 is involved in the progression of several cancers, including breast and lung cancer [
77]. The increased proline synthesis is linked to increased production and stiffness of the ECM and plays an important role in tumourigenesis [
78].
7. ECM in Solid Cancers
Tumour progression requires continuous interaction between the ECM and tumour cells where increased cytoskeleton signalling reduces the number of intercellular adhesion molecules, induces mesenchymal transition, upregulates membrane integrins, enhances the migratory potential [
7] and therapeutic resistance [
67]. Escalated deposition and cross-linking of collagen interfere with cell polarity, cell adhesion, and integrin signalling and promote tumour progression. ECM degradation and loss of BM integrity due to MMP are hallmarks of invasive lesions [
79]. Another cancer hallmark is the tumour cell production of glycoproteins and proteoglycans with altered glycosylation (e.g., cancer antigen 19-9 and 125, carcinoembryonic antigen, prostate-specific antigen and alpha-fetoprotein) secreted or shed from the cell membranes into the bloodstream where they may serve as tumour-associated biomarkers) [
80].
ECM—the largest part of a solid tumour—applies mechanical and non-mechanical forces on tumour tissue, such as stiffening, increased interstitial fluid pressure, collapsing vessels, hypoxia, and acidity [
81] and compromises the outcome of oncological treatments and prognosis [
82].
In melanoma and breast cancer, increased amounts of collagen I correlated with disease progression and reduced survival [
83,
84,
85]. Further, collagen and fibronectin are involved in angiogenesis. Interacting with α1β1, α2β1, ανβ3, and ανβ5 integrins, collagen I activates mitogen-activated protein kinase (MAPK) pathways supporting the survival of endothelial cells, remodelling the actin cytoskeleton, and influencing the cells to form lumens [
86]. Fibronectin plays a pivotal role in the assembly of vascular matrix components [
87], while MMP releases GF, VEGF, and chemokines bound within the ECM [
88,
89] to remodel the ECM and direct endothelial migration and capillary movements along aligned ECM fibres [
90,
91].
When nutrient levels run low, integrin-bound glycoproteins of the ECM are easily endocytosed and internalised into lysosomes by tumour cells. The role of ECM as a nutrient provider represents a potential therapeutic target to inhibit integrin uptake in stromal-enriched cancers [
92]. Ligand-bound integrin trafficking has recently been shown to affect nutrient signalling through the rapamycin (mTOR) signalling pathway [
92,
93]. With sufficient nutrient availability, mTOR induces anabolic processes such as protein, nucleotide, and lipid biosynthesis and inhibits lysosomal biogenesis and cellular autophagy [
94] through two independent complexes of mTOR. The complex mTORC1 adjusts cell growth and proliferation in response to GF and amino acids, while mTORC2 is involved in actin organisation and cell proliferation and survival [
95]. During nutrient starvation, the activity of mTORC1 is downregulated, allowing cells to use other sources of nutrient acquisition, such as autophagy [
96,
97].
The ECM-attached cells induce adaptive responses or compensatory homeostatic feedback loops, leading to the induction of several pro-survival proteins, including receptor tyrosine kinases and antiapoptotic proteins including the activation of MAPK, PI3K, AKT and the human epidermal growth factor 3 (HER3) [
98].
The alignment of collagen fibres by lysyl oxidase (LOX) produced by cancer cells and CAF directs cancer cell migration and induces their proliferation [
99,
100,
101]. In an in vitro study on benign and malignant human ovarian cell lines, the ability to rapidly remodel the matrix enabled tumour cell migration along aligned fibres and changed direction according to microenvironmental cues [
102].
The ECM is a storage facility for nutrients, proteases, morphogens, and GF that create pro-migratory gradients. These molecules are released by proteolytic degradation of the ECM, which regulates the rate and intensity of migration [
103]. Due to the ongoing modulation of the ECM, the TME is rich in protease-digested fragments that may influence the metastatic potential and apoptosis of cancer cells [
104]. These fragments may even display opposite effects as compared to their molecules of origin, making studies on the TME even more challenging [
105].
Notably, the binding of an ECM-associated GF to its receptor (GFR) may prevent its endocytosis and secure prolonged, upregulated signalling by the ECM-GF/GFR complex [
106]. In addition, cells may generate different responses to the same effector molecule bound to different matrix molecules under the same GF conditions [
107].