Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Stroke is one of the leading causes of morbidity and mortality worldwide. A main cause of brain damage by stroke is ischemia-reperfusion (IR) injury due to the increased production of reactive oxygen species (ROS) and energy failure caused by changes in mitochondrial metabolism. Reverse electron transfer (RET) has been implicated in excessive ROS production during IR injury in stroke and new insights into the mechanism of RET gained from various systems in recent studies will help understand stroke pathophysiology and inform therapy.
- mitochondrial complex I
- reverse electron transport (RET)
- reactive oxygen species (ROS)
1. Introduction
Stroke is the second leading cause of morbidity and mortality worldwideafter cancer. In almost 80% of the cases, stroke develops due to a cerebral artery obstruction and/or occlusion [1][2]. During ischemic stroke, the absence of blood supply deprives the brain cells of the glucose and oxygen nutrients they require, disrupting their cellular homeostasis and ultimately resulting in cell death [2]. Mitochondrial dysfunction and deleterious post-stroke ROS are considered hallmark stroke pathologies [3]. A number of pathophysiological processes, including oxidative stress, excitotoxicity, dysregulated endocrine signaling, inflammation, and apoptosis, are involved in the complex pathophysiological process known as cerebral ischemia/reperfusion injury (CIRI), which frequently causes neuronal injury, cell death, and permanent brain damage [3]. ROS-induced oxidative stress during cerebral ischemia leads to eventual cell death after reperfusion. Mitochondrial ROS (mito-ROS) plays a detrimental role in neuronal death during CIRI at several key stages: inflammation, blood brain barrier (BBB) disruption, mitochondrial respiratory chain complex I-III dysfunction, oedema formation, and apoptosis and autophagy [4][5][6] Currently, thrombolytic and endoscopic thrombectomy are the only therapeutic options, along with post-stroke conservative treatments. The tissue plasminogen activator (tPA) is the only Federal Drug Administration (FDA)-approved therapy for stroke treatment, but due to its various side effects and limited therapeutic window, it only benefits a small portion of stroke patients [7][8][9][10][11]. Hence, there is an urgent need to develop safer and novel therapeutic options for the treatment of ischemia reperfusion injuries. Due to reduced blood flow and hypoxic conditions, mitochondria are heavily affected by the low O2 and glucose environment. Since the 1960s, the effect of ischemia reperfusion injury on mitochondria has been a research focus and extensive efforts have been dedicated to developing therapeutic strategies to combat reperfusion injury.
2. ROS and Oxidative Stress in Stroke
2.1. ROS Generation during Stroke
Oxidative stress is the result of an imbalance between ROS production and antioxidant defense mechanisms in cells [3]. Because of its high and specific metabolic activity, neurons are particularly vulnerable to oxidative damage. High oxygen consumption, almost entirely oxidative phosphorylation, low energy reserves, high concentrations of peroxidizable lipids, and high levels of iron acting as prooxidants all contribute to this vulnerability [12]. As a result, neuronal cells are extremely vulnerable to metabolic/ischemic damage and the associated oxidative stress [13]. The macro- and micromolecular changes of neuronal cells ends in neurodegeneration in various neurological disorders, e.g., post-stroke dementia, Alzheimer’s disease (AD), vascular dementia, and others [14]. The progression of ischemic stroke pathology is closely connected to dysregulated ROS. The most common ROS are superoxide (O2−), hydrogen peroxide (H2O2), hydroxyl radical (HO−), hypochlorous acid (HOCl−), nitric oxide (NO), and peroxynitrite (ONOO−) that are produced by either intracellular responses (mitochondria) or extracellular inflammation [3][15]. The intracellular production of ROS is mainly due to the altered metabolic activity of the mitochondrial respiratory chain, whereas extracellularly it is a result of inflammasome activation and the immune response [16][17]. These reactive molecules can cause lipid peroxidation, protein oxidation, and DNA and RNA damage, resulting in cellular homeostatic failure and tissue damage (Figure 1). Nitric oxide synthetase (NOS) produces NO that is instrumental in the immune response; phagocytes produce large amounts of NO during ischemic brain injury [3][18]. There are several natural defense mechanisms in the cell to remove ROS or prevent oxidative damage [19]. These include catalase (CAT), heme oxygenase (HO), glutathione reductase, glutathione (GSH), glutathione peroxidase (GSH-Px), superoxide dismutase (SOD), and Vitamins E and C. Microglia and astrocytes are the primary producers of ROS and reactive nitrogen species (RNS), the latter is produced by endothelial NOS (eNOS), inducible NOS (iNOS), or neuronal NOS (nNOS) during ischemic brain injury, which together influence synaptic transmission as well as non-synaptic communication between neurons and glia [20][21]. ROS and RNS diffuse to the oligodendrocyte myelin sheath during periods of increased neuronal activity, activating the protein kinase C and post-translationally modifying the myelin basic protein, a key structural component of myelin [22][23].
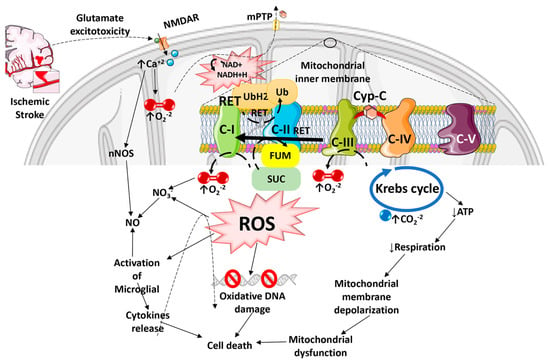
Figure 1. Diagram illustrating how Reactive Oxygen Species especially Reactive Oxygen Species generated by the Reverse Electron Transport process, and the ensuing oxidative stress contributes to mitochondrial dysfunction, cell death, and progressive ischemic stroke pathology. ROS can damage mitochondrial DNA because of the lack of a chromatin-like structure that would protect DNA against ROS insults. ROS can also damage lipids and protein structures in mitochondrial matrix and further exacerbate mitochondrial dysfunction. Mitochondrial ROS released into the cytosol can activate microglia and astrocytes, causing neuroinflammation and death of injured neurons.
2.2. ROS-Induced Damages during Stroke
The consequences of ROS imbalance in ischemic stroke are significant, and include apoptosis, disruption of the BBB, inflammation, edema formation, autophagy, and other pathophysiological events. O2, H2O2, and NO play critical roles in neuron-glia communication in the hippocampus [24]. The synaptic long-term potentiation (LTP), which is necessary for memory formation in the hippocampus, also becomes impaired by excessive ROS production during ischemia, resulting in post-stroke cognitive decline [25][26]. ROS-mediated injury can result in the formation of conjugated dienic hydroperoxides, which can be degraded into aldehydes, dienals, or alkanes that are extremely toxic to neurons and white matter, resulting in apoptosis followed by chronic neurodegeneration after ischemic brain damage [27]. The inflammation and oxidative stress that develop in the brain after a stroke have been linked more recently to tryptophan oxidation via the kynurenine pathway [28]. The release of glutamate is a crucial mechanism that determines tissue damage after cerebral ischemia [29]. On the other hand, when the a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are activated, oxygen is produced. When oxygen combines with NO, it creates the extremely harmful ONOO. When one of the ROS species is inactivated, the vessel can expand due to the other reactant (ROS) escaping [30]. It has also been shown that increased kynurenic acid levels are linked to poorer outcomes, and the infarct volume is strongly correlated with a decreased ratio of 3-hydroxyanthranilic acid to anthranilic acid (a free radical generator) [31][32]. Nuclear DNA damage has been linked to two distinct mechanisms, including DNA fragmentation caused by endonucleases and oxidative modification. In ischemic stroke injury, poly (ADP-ribose) polymerase (PARP) activation occurs in two phases, starting in the neuronal components and localizing 3–4 days later in the infiltrating inflammatory cells [33]. It has also been suggested that there may be reductions in the nuclear protein apurinic apyrimidinic endonuclease (APE/Ref-1), which cleaves ROS-induced apyrimidinic sites in oxidized DNA [27]. Moreover, following transient global ischemia, the p53-upregulated modulator of apoptosis (PUMA) is upregulated in the hippocampal neurons [34]. ROS can also activate caspase-activated DNase (CAD), which cleaves DNA and causes apoptosis, resulting in progressive neurodegeneration and associated post-stroke disorders [35]. Thus, excessive ROS production can cause mitochondrial dysfunction leading to overall cellular dysfunction and cell death in ischemic stroke (Figure 1).
This entry is adapted from the peer-reviewed paper 10.3390/antiox12040895
References
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480.
- Owolabi, M.O.; Thrift, A.G.; Mahal, A.; Ishida, M.; Martins, S.; Johnson, W.D.; Pandian, J.; Abd-Allah, F.; Yaria, J.; Phan, H.T.; et al. Primary stroke prevention worldwide: Translating evidence into action. Lancet Public Health 2022, 7, e74–e85.
- Chavda, V.; Chaurasia, B.; Garg, K.; Deora, H.; Umana, G.E.; Palmisciano, P.; Scalia, G.; Lu, B. Molecular mechanisms of oxidative stress in stroke and cancer. Brain Disord. 2022, 5, 100029.
- Monsour, M.; Borlongan, C.V. The central role of peripheral inflammation in ischemic stroke. J. Cereb. Blood Flow Metab. 2023.
- Carinci, M.; Vezzani, B.; Patergnani, S.; Ludewig, P.; Lessmann, K.; Magnus, T.; Casetta, I.; Pugliatti, M.; Pinton, P.; Giorgi, C. Different roles of mitochondriain cell death and inflammation: Focusing on mitochondrial quality control in ischemic stroke and reperfusion. Biomedicines 2021, 9, 169.
- Sarmah, D.; Kaur, H.; Saraf, J.; Vats, K.; Pravalika, K.; Wanve, M.; Kalia, K.; Borah, A.; Kumar, A.; Wang, X.; et al. Mitochondrial dysfunction in stroke: Implications of stem cell therapy. Transl. Stroke Res. 2019, 10, 121–136.
- Simão, F.; Ustunkaya, T.; Clermont, A.C.; Feener, E.P. Plasma kallikrein mediates brain hemorrhage and edema caused by tissue plasminogen activator therapy in mice after stroke. Blood J. Am. Soc. Hematol. 2017, 129, 2280–2290.
- Saver, J.L.; Goyal, M.; Bonafe, A.; Diener, H.C.; Levy, E.I.; Pereira, V.M.; Albers, G.W.; Cognard, C.; Cohen, D.J.; Hacke, W.; et al. Solitaire™ with the Intention for Thrombectomy as Primary Endovascular Treatment for Acute Ischemic Stroke (SWIFTPRIME) trial: Protocol for a randomized, controlled, multicenter study comparing the Solitaire revascularization device with IV tPA with IV tPA alone in acute ischemic stroke. Int. J. Stroke 2015, 10, 439–448.
- Grossberg, J.A.; Rebello, L.C.; Haussen, D.C.; Bouslama, M.; Bowen, M.; Barreira, C.M.; Belagaje, S.R.; Frankel, M.R.; Nogueira, R.G. Beyond large vessel occlusion strokes: Distal occlusion thrombectomy. Stroke 2018, 49, 1662–1668.
- Albers, G.W.; Clark, W.M.; Madden, K.P.; Hamilton, S.A. ATLANTIS trial: Results for patients treated within 3 hours of stroke onset. Stroke 2002, 33, 493–496.
- Jadhav, A.P.; Desai, S.M.; Kenmuir, C.L.; Rocha, M.; Starr, M.T.; Molyneaux, B.J.; Gross, B.A.; Jankowitz, B.T.; Jovin, T.G. Eligibility for endovascular trial enrollment in the 6-to-24 hour time window: Analysis of a single comprehensive stroke center. Stroke 2018, 49, 1015–1017.
- Martynov, M.Y.; Zhuravleva, M.V.; Vasyukova, N.S.; Kuznetsova, E.V.; Kameneva, T.R. Oxidative stress in the pathogenesis of stroke and its correction. Zhurnal Nevrol. Psikhiatrii Im. Korsakova 2023, 123, 16–27.
- Olufunmilayo, E.O.; Gerke Duncan, M.B.; Holsinger, R.D. Oxidative Stress and Antioxidants in Neurodegenerative Disorders. Antioxidants 2023, 12, 517.
- Chan, M.K.; Nalapko, Y. Ageing brain and neurodegeneration: Preventive and regenerative medicine. In Handbook of Anti-Aging Medicine; European Wellness Academy: London, UK, 2023.
- Shehjar, F.; Maktabi, B.; Rahman, Z.A.; Bahader, G.A.; James, A.W.; Naqvi, A.; Mahajan, R.; Shah, Z.A. Stroke: Molecular mechanisms and therapies: Update on recent developments. Neurochem. Int. 2023, 162, 105458.
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110.
- Danieli, M.G.; Antonelli, E.; Piga, M.A.; Cozzi, M.F.; Allegra, A.; Gangemi, S. Oxidative stress, mitochondrial dysfunction, and respiratory chain enzyme defects in inflammatory myopathies. Autoimmun. Rev. 2023, 21, 103308.
- García-Sánchez, A.; Miranda-Díaz, A.G.; Cardona-Muñoz, E.G. The role of oxidative stress in physiopathology and pharmacological treatment with pro-and antioxidant properties in chronic diseases. Oxidative Med. Cell. Longev. 2020, 2020, 2082145.
- Roleira, F.M.; Tavares-da-Silva, E.J.; Garrido, J.; Borges, F. Antioxidants and stroke: Success and pitfalls. Transl. Stroke Res. Target Sel. Clin. Trials 2012, 3, 117–143.
- Shirley, R.; Ord, E.N.; Work, L.M. Oxidative stress and the use of antioxidants in stroke. Antioxidants 2014, 3, 472–501.
- Ascherio, A. Antioxidants and stroke. Am. J. Clin. Nutr. 2000, 72, 337–338.
- Zhao, S.C.; Ma, L.S.; Chu, Z.H.; Xu, H.; Wu, W.Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol. 2017, 38, 445–458.
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system:A mixed blessing. Cell Prolif. 2020, 53, e12781.
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative stress-mediated blood-brain barrier (BBB) disruption in neurological diseases. Oxidative Med. Cell. Longev. 2020, 4356386.
- Zia, A.; Pourbagher-Shahri, A.M.; Farkhondeh, T.; Samarghandian, S. Molecular and cellular pathways contributing to brain aging. Behav. Brain Funct. 2021, 17, 6.
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011, 14, 2013–2054.
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 2009, 4, 461–470.
- Brouns, R.; Verkerk, R.; Aerts, T.; DeSurgeloose, D.; Wauters, A.; Scharpé, S.; DeDeyn, P.P. The role of tryptophan catabolism along the kynurenine pathway in acute ischemic stroke. Neurochem. Res. 2010, 35, 1315–1322.
- Hazell, A.S. Excitotoxic mechanisms in stroke: An update of concepts and treatment strategies. Neurochem. Int. 2007, 50, 941–953.
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537.
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2002, 1, 609–620.
- Darlington, L.G.; Mackay, G.M.; Forrest, C.M.; Stoy, N.; George, C.; Stone, T.W. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur. J. Neurosci. 2007, 26, 2211–2221.
- Liu, S.; Luo, W.; Wang, Y. Emerging role of PARP-1and PARthanatos in ischemic stroke. J. Neurochem. 2022, 160, 74–87.
- Niizuma, K.; Endo, H.; Nito, C.; Myer, D.J.; Chan, P.H. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke 2009, 40, 618–625.
- Saeed, S.A.; Shad, K.F.; Saleem, T.; Javed, F.; Khan, M.U. Some new prospects in the understanding of the molecular basis of the pathogenesis of stroke. Exp. Brain Res. 2007, 182, 1–10.
This entry is offline, you can click here to edit this entry!