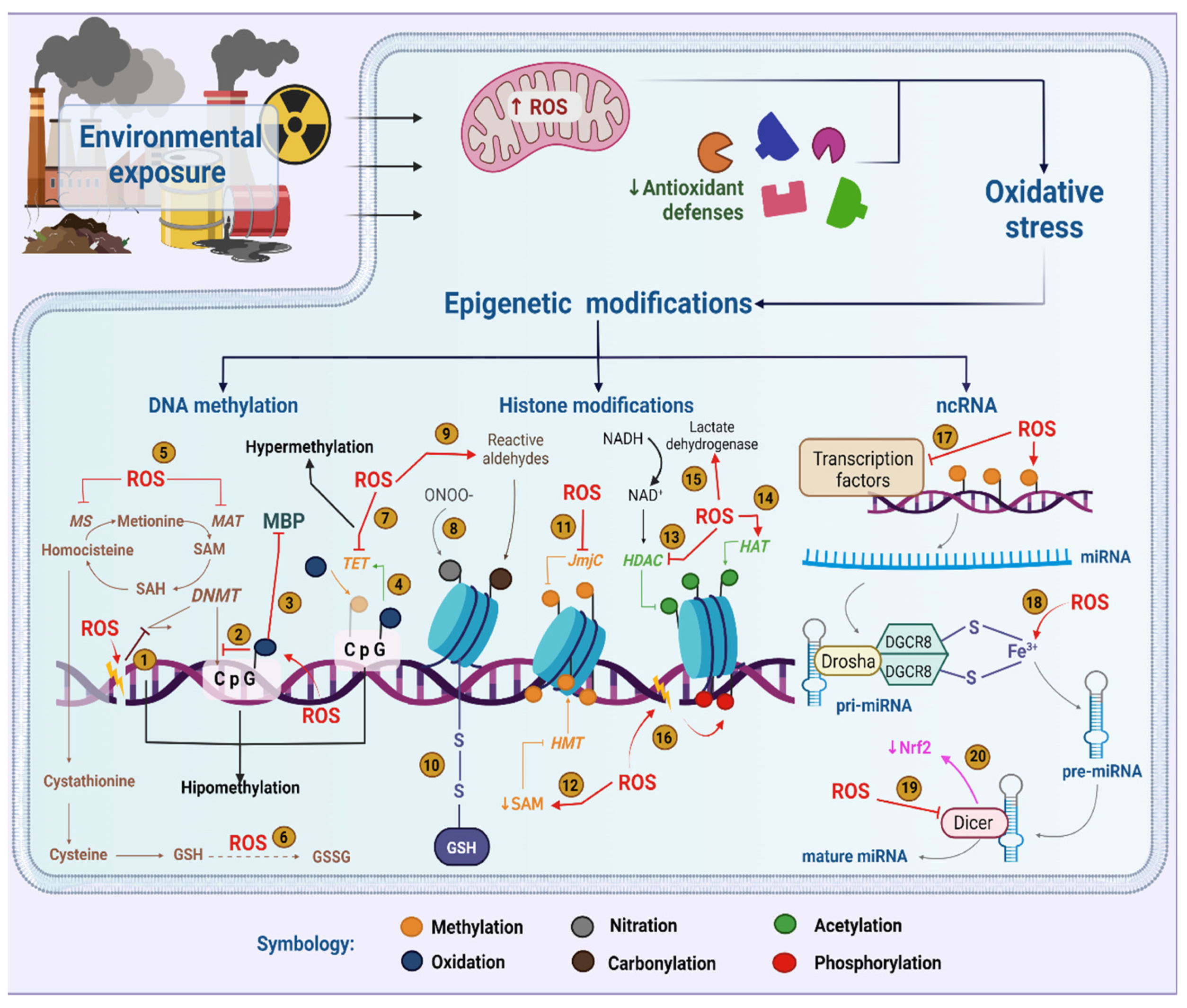
Complex molecular mechanisms define our responses to environmental stimuli. Beyond the DNA sequence itself, epigenetic machinery orchestrates changes in gene expression induced by diet, physical activity, stress and pollution, among others. Importantly, nutrition has a strong impact on epigenetic players and, consequently, sustains a promising role in the regulation of cellular responses such as oxidative stress. As oxidative stress is a natural physiological process where the presence of reactive oxygen-derived species and nitrogen-derived species overcomes the uptake strategy of antioxidant defenses, it plays an essential role in epigenetic changes induced by environmental pollutants and culminates in signaling the disruption of redox control.
- nutrition
- 2D culture
- epigenetics
- extracellular vesicles
1. Introduction
2. Epigenetics in the Context of Environmental Exposure
3. Relationship between Epigenetics and Oxidative Stress in the Context of Environmental Exposure
4. Microenvironment and Nutritional Influence on the Preservation of Epigenetic Marks
5. Effects of Dietary Nutrients on the Epigenome
This entry is adapted from the peer-reviewed paper 10.3390/antiox12030771
References
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638.
- Skinner, M.K. Environmental Epigenetics and a Unified Theory of the Molecular Aspects of Evolution: A Neo-Lamarckian Concept that Facilitates Neo-Darwinian Evolution. Genome Biol. Evol. 2015, 7, 1296–1302.
- Xu, Y.; Doonan, S.R.; Ordog, T.; Bailey, R.C. Translational Opportunities for Microfluidic Technologies to Enable Precision Epigenomics. Anal. Chem. 2020, 92, 7989–7997.
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707.
- Rubio, K.; Dobersch, S.; Barreto, G. Functional interactions between scaffold proteins, noncoding RNAs, and genome loci induce liquid-liquid phase separation as organizing principle for 3-dimensional nuclear architecture: Implications in cancer. FASEB J. 2019, 33, 5814–5822.
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304.
- Singh, I.; Contreras, A.; Cordero, J.; Rubio, K.; Dobersch, S.; Günther, S.; Jeratsch, S.; Mehta, A.; Krüger, M.; Graumann, J.; et al. MiCEE is a ncRNA-protein complex that mediates epigenetic silencing and nucleolar organization. Nat. Genet. 2018, 50, 990–1001.
- Dobersch, S.; Rubio, K.; Singh, I.; Günther, S.; Graumann, J.; Cordero, J.; Castillo-Negrete, R.; Huynh, M.B.; Mehta, A.; Braubach, P.; et al. Positioning of nucleosomes containing gamma-H2AX precedes active DNA demethylation and transcription initiation. Nat. Commun. 2021, 12, 1072.
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022.
- Gardiner-Garden, M.; Frommer, M. CpG Islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282.
- Barreto, G.; Schäfer, A.; Marhold, J.; Stach, D.; Swaminathan, S.K.; Handa, V.; Döderlein, G.; Maltry, N.; Wu, W.; Lyko, F.; et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 2007, 445, 671–675.
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750.
- Meehan, R.R.; Thomson, J.P.; Lentini, A.; Nestor, C.E.; Pennings, S. DNA methylation as a genomic marker of exposure to chemical and environmental agents. Curr. Opin. Chem. Biol. 2018, 45, 48–56.
- Mehta, A.; Dobersch, S.; Romero-Olmedo, A.J.; Barreto, G. Epigenetics in lung cancer diagnosis and therapy. Cancer Metastasis Rev. 2015, 34, 229–241.
- Martin, E.M.; Fry, R.C. Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations. Annu. Rev. Public Health 2018, 39, 309–333.
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874.
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492.
- Thomson, J.P.; Meehan, R.R. The application of genome-wide 5-hydroxymethylcytosine studies in cancer research. Epigenomics 2017, 9, 77–91.
- Thomson, J.P.; Ottaviano, R.; Unterberger, E.B.; Lempiäinen, H.; Muller, A.; Terranova, R.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; Lyall, M.J.; et al. Loss of Tet1-Associated 5-Hydroxymethylcytosine Is Concomitant with Aberrant Promoter Hypermethylation in Liver Cancer. Cancer Res. 2016, 76, 3097–3108.
- Ozturk, N.; Singh, I.; Mehta, A.; Braun, T.; Barreto, G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front. Cell Dev. Biol. 2014, 2, 5.
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260.
- Wang, Y.; Fischle, W.; Cheung, W.; Jacobs, S.; Khorasanizadeh, S.; Allis, C.D. Beyond the double helix: Writing and reading the histone code. Novartis Found. Symp. 2004, 259, 3–17, discussion 17-21, 163–9.
- Singh, I.; Ozturk, N.; Cordero, J.; Mehta, A.; Hasan, D.; Cosentino, C.; Sebastian, C.; Krüger, M.; Looso, M.; Carraro, G.; et al. High mobility group protein-mediated transcription requires DNA damage marker gamma-H2AX. Cell Res. 2015, 25, 837–850.
- Zhang, M.; Zhao, J.; Lv, Y.; Wang, W.; Feng, C.; Zou, W.; Su, L.; Jiao, J. Histone Variants and Histone Modifications in Neurogenesis. Trends Cell Biol. 2020, 30, 869–880.
- Nakayama, T.; Takami, Y. Participation of Histones and Histone-Modifying Enzymes in Cell Functions through Alterations in Chromatin Structure. J. Biochem. 2001, 129, 491–499.
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281.
- Stillman, B. Histone Modifications: Insights into Their Influence on Gene Expression. Cell 2018, 175, 6–9.
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The Effects of Cadmium Toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782.
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119.
- Wang, L.; Gao, Y.; Zheng, X.; Liu, C.; Dong, S.; Li, R.; Zhang, G.; Wei, Y.; Qu, H.; Li, Y.; et al. Histone Modifications Regulate Chromatin Compartmentalization by Contributing to a Phase Separation Mechanism. Mol. Cell 2019, 76, 646–659.e6.
- Johnson, M.O.; Siska, P.J.; Contreras, D.C.; Rathmell, J.C. Nutrients and the microenvironment to feed a T cell army. Semin. Immunol. 2016, 28, 505–513.
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Padilla, P.G.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873.
- Niculescu, M.D.; Zeisel, S.H. Diet, Methyl Donors and DNA Methylation: Interactions between Dietary Folate, Methionine and Choline. J. Nutr. 2002, 132, S2333–S2335.
- Twinn, D.; Hjort, L.; Novakovic, B.; Ozanne, S.; Saffery, R. Intrauterine programming of obesity and type 2 diabetes. Diabetologia 2019, 62, 1789–1801.
- Caudron-Herger, M.; Rippe, K. Nuclear architecture by RNA. Curr. Opin. Genet. Dev. 2012, 22, 179–187.
- Rubio, K.; Castillo-Negrete, R.; Barreto, G. Non-coding RNAs and nuclear architecture during epithelial-mesenchymal transition in lung cancer and idiopathic pulmonary fibrosis. Cell. Signal. 2020, 70, 109593.
- Zhang, W.; Dolan, M.E. Beyond the HapMap Genotypic Data: Prospects of Deep Resequencing Projects. Curr. Bioinform. 2008, 3, 178.
- Vojtech, L.; Woo, S.; Hughes, S.; Levy, C.; Ballweber, L.; Sauteraud, R.P.; Strobl, J.; Westerberg, K.; Gottardo, R.; Tewari, M.; et al. Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic Acids Res. 2014, 42, 7290–7304.
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114.
- Park, C.W.; Zeng, Y.; Zhang, X.; Subramanian, S.; Steer, C.J. Mature microRNAs identified in highly purified nuclei from HCT116 colon cancer cells. RNA Biol. 2010, 7, 606–614.
- Leucci, E.; Patella, F.; Waage, J.; Holmstrøm, K.; Lindow, M.; Porse, B.; Kauppinen, S.; Lund, A.H. microRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci. Rep. 2013, 3, 2535.
- Politz, J.C.R.; Zhang, F.; Pederson, T. MicroRNA-206 colocalizes with ribosome-rich regions in both the nucleolus and cytoplasm of rat myogenic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 18957–18962.
- Reyes-Gutierrez, P.; Politz, J.C.R.; Pederson, T. A mRNA and Cognate MicroRNAs Localize in the Nucleolus. Nucleus 2014, 5, 636–642.
- Rubio, K.; Singh, I.; Dobersch, S.; Sarvari, P.; Günther, S.; Cordero, J.; Mehta, A.; Wujak, L.; Cabrera-Fuentes, H.; Chao, C.-M.; et al. Inactivation of nuclear histone deacetylases by EP300 disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 2229.
- Schmitz, K.-M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010, 24, 2264–2269.
- Yang, L.; Lin, C.; Liu, W.; Zhang, J.; Ohgi, K.A.; Grinstein, J.D.; Dorrestein, P.C.; Rosenfeld, M.G. ncRNA- and Pc2 Methylation-Dependent Gene Relocation between Nuclear Structures Mediates Gene Activation Programs. Cell 2011, 147, 773–788.
- Hu, X.; Feng, Y.; Zhang, D.; Zhao, S.D.; Hu, Z.; Greshock, J.; Zhang, Y.; Yang, L.; Zhong, X.; Wang, L.-P.; et al. A Functional Genomic Approach Identifies FAL1 as an Oncogenic Long Noncoding RNA that Associates with BMI1 and Represses p21 Expression in Cancer. Cancer Cell 2014, 26, 344–357.
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674.
- Wallace, D.R.; Taalab, Y.M.; Heinze, S.; Lovaković, B.T.; Pizent, A.; Renieri, E.; Tsatsakis, A.; Farooqi, A.A.; Javorac, D.; Andjelkovic, M.; et al. Toxic-Metal-Induced Alteration in miRNA Expression Profile as a Proposed Mechanism for Disease Development. Cells 2020, 9, 901.
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 2013, 19, 1141–1146.
- Martinez-Zamudio, R.; Ha, H.C. Environmental epigenetics in metal exposure. Epigenetics 2011, 6, 820–827.
- Menezo, Y.J.; Dale, B.; Elder, K. The negative impact of the environment on methylation/epigenetic marking in gametes and embryos: A plea for action to protect the fertility of future generations. Mol. Reprod. Dev. 2019, 86, 1273–1282.
- Samet, J.M.; Wages, P. Oxidative stress from environmental exposures. Curr. Opin. Toxicol. 2018, 7, 60–66.
- Miguel, V.; Lamas, S.; Espinosa-Diez, C. Role of non-coding-RNAs in response to environmental stressors and consequences on human health. Redox Biol. 2020, 37, 101580.
- Bhargava, A.; Shukla, A.; Bunkar, N.; Shandilya, R.; Lodhi, L.; Kumari, R.; Gupta, P.K.; Rahman, A.; Chaudhury, K.; Tiwari, R.; et al. Exposure to ultrafine particulate matter induces NF-kappabeta mediated epigenetic modifications. Environ. Pollut. 2019, 252 Pt A, 39–50.
- Soberanes, S.; Gonzalez, A.; Urich, D.; Chiarella, S.E.; Radigan, K.A.; Osornio-Vargas, A.; Joseph, J.; Kalyanaraman, B.; Ridge, K.M.; Chandel, N.S.; et al. Particulate matter Air Pollution induces hypermethylation of the p16 promoter Via a mitochondrial ROS-JNK-DNMT1 pathway. Sci. Rep. 2012, 2, 275.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Zhou, Y.; Shen, S.; Du, C.; Wang, Y.; Liu, Y.; He, Q. A role for the mitotic proteins Bub3 and BuGZ in transcriptional regulation of catalase-3 expression. PLoS Genet. 2022, 18, e1010254.
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167.
- Branca, J.J.V.; Pacini, A.; Gulisano, M.; Taddei, N.; Fiorillo, C.; Becatti, M. Cadmium-Induced Cytotoxicity: Effects on Mitochondrial Electron Transport Chain. Front. Cell Dev. Biol. 2020, 8, 604377.
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264.
- Ralph, S.J.; Moreno-Sanchez, R.; Neuzil, J.; Rodriguez-Enriquez, S. Inhibitors of succinate: Quinone reductase/Complex II regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm. Res. 2011, 28, 2695–2730.
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698.
- Dijkstra, G.; Moshage, H.; Van Dullemen, H.M.; De Jager-Krikken, A.; Tiebosch, A.T.M.G.; Kleibeuker, J.H.; Jansen, P.L.M.; van Goor, H. Expression of nitric oxide synthases and formation of nitrotyrosine and reactive oxygen species in inflammatory bowel disease. J. Pathol. 1998, 186, 416–421.
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201.
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84.
- Rubin, H. Dynamics of cell transformation in culture and its significance for tumor development in animals. Proc. Natl. Acad. Sci. USA 2017, 114, 12237–12242.
- Ou, K.; Hamo, D.; Schulze, A.; Roemhild, A.; Kaiser, D.; Gasparoni, G.; Salhab, A.; Zarrinrad, G.; Amini, L.; Schlickeiser, S.; et al. Strong Expansion of Human Regulatory T Cells for Adoptive Cell Therapy Results in Epigenetic Changes Which May Impact Their Survival and Function. Front. Cell Dev. Biol. 2021, 9, 751590.
- Cox, M.C.; Deng, C.; Naler, L.B.; Lu, C.; Verbridge, S.S. Effects of Culture Condition on Epigenomic Profiles of Brain Tumor Cells. ACS Biomater. Sci. Eng. 2019, 5, 1544–1552.
- Weissbein, U.; Plotnik, O.; Vershkov, D.; Benvenisty, N. Culture-induced recurrent epigenetic aberrations in human pluripotent stem cells. PLoS Genet. 2017, 13, e1006979.
- Nazor, K.L.; Altun, G.; Lynch, C.; Tran, H.; Harness, J.V.; Slavin, I.; Garitaonandia, I.; Müller, F.-J.; Wang, Y.-C.; Boscolo, F.S.; et al. Recurrent Variations in DNA Methylation in Human Pluripotent Stem Cells and Their Differentiated Derivatives. Cell Stem Cell 2012, 10, 620–634.
- Mallon, B.S.; Chenoweth, J.G.; Johnson, K.R.; Hamilton, R.S.; Tesar, P.J.; Yavatkar, A.S.; Tyson, L.J.; Park, K.; Chen, K.G.; Fann, Y.C.; et al. StemCellDB: The Human Pluripotent Stem Cell Database at the National Institutes of Health. Stem Cell Res. 2013, 10, 57–66.
- Fareed, M.M.; Shityakov, S.; Ullah, S.; Qasmi, M. The Role of Vitamins in DNA Methylation as Dietary Supplements or Neutraceuticals: A systematic review. Curr. Mol. Med. 2022.
- Pehlivan, F.E. Vitamin C: An Epigenetic Regulator. In Vitamin C—An Update on Current Uses and Functions; IntechOpen Limited: London, UK, 2019.
- Bornstein, M.H. Attention in infancy and the prediction of cognitive capacities in childhood. Semin. Perinatol. 1989, 13, 450–457.
- Guz, J.; Oliński, R. The role of vitamin C in epigenetic regulation. Postep. Hig. Med. Dosw. 2017, 71, 747–760.
- Camarena, V.; Wang, G. The epigenetic role of vitamin C in health and disease. Cell. Mol. Life Sci. 2016, 73, 1645–1658.
- Brabson, J.P.; Leesang, T.; Mohammad, S.; Cimmino, L. Epigenetic Regulation of Genomic Stability by Vitamin C. Front. Genet. 2021, 12, 675780.
- Hore, T.A.; von Meyenn, F.; Ravichandran, M.; Bachman, M.; Ficz, G.; Oxley, D.; Santos, F.; Balasubramanian, S.; Jurkowski, T.P.; Reik, W. Retinol and ascorbate drive erasure of epigenetic memory and enhance reprogramming to naïve pluripotency by complementary mechanisms. Proc. Natl. Acad. Sci. USA 2016, 113, 12202–12207.
- Urvalek, A.; Laursen, K.B.; Gudas, L.J. The Roles of Retinoic Acid and Retinoic Acid Receptors in Inducing Epigenetic Changes. Subcell. Biochem. 2014, 70, 129–149.
- Angrisano, T.; Sacchetti, S.; Natale, F.; Cerrato, A.; Pero, R.; Keller, S.; Peluso, S.; Perillo, B.; Avvedimento, V.E.; Fusco, A.; et al. Chromatin and DNA methylation dynamics during retinoic acid-induced RET gene transcriptional activation in neuroblastoma cells. Nucleic Acids Res. 2011, 39, 1993–2006.
- Uekawa, A.; Katsushima, K.; Ogata, A.; Kawata, T.; Maeda, N.; Kobayashi, K.-I.; Maekawa, A.; Tadokoro, T.; Yamamoto, Y. Change of epigenetic control of cystathionine beta-synthase gene expression through dietary vitamin B12 is not recovered by methionine supplementation. J. Nutrigenet. Nutr. 2009, 2, 29–36.
- Mahajan, A.; Sapehia, D.; Thakur, S.; Mohanraj, P.S.; Bagga, R.; Kaur, J. Effect of imbalance in folate and vitamin B12 in maternal/parental diet on global methylation and regulatory miRNAs. Sci. Rep. 2019, 9, 17602.
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and Human Health: Lessons from Vitamin D Receptor Null Mice. Endocr. Rev. 2008, 29, 726–776.
- Karlic, H.; Varga, F. Impact of vitamin D metabolism on clinical epigenetics. Clin. Epigenetics 2011, 2, 55–61.
- Zhu, H.; Wang, X.; Shi, H.; Su, S.; Harshfield, G.A.; Gutin, B.; Snieder, H.; Dong, Y. A Genome-Wide Methylation Study of Severe Vitamin D Deficiency in African American Adolescents. J. Pediatr. 2013, 162, 1004–1009.e1.
- Bikle, D.D. Vitamin D regulation of and by long non coding RNAs. Mol. Cell. Endocrinol. 2021, 532, 111317.
- Luna, R.C.; dos Santos Nunes, M.K.; Monteiro, M.G.; da Silva, C.S.; do Nascimento, R.A.; Lima, R.P.; Pimenta, F.C.; de Oliveira, N.F.; Persuhn, D.C.; de Almeida, A.T.; et al. α-Tocopherol influences glycaemic control and miR-9-3 DNA methylation in overweight and obese women under an energy-restricted diet: A randomized, double-blind, exploratory, controlled clinical trial. Nutr. Metab. 2018, 15, 49.
- Shankar, S.; Kumar, D.; Srivastava, R.K. Epigenetic modifications by dietary phytochemicals: Implications for personalized nutrition. Pharmacol. Ther. 2013, 138, 1–17.
- Yu, X.-D.; Guo, Z.S. Epigenetic drugs for cancer treatment and prevention: Mechanisms of action. Biomol. Concepts 2010, 1, 239–251.
- Cione, E.; La Torre, C.; Cannataro, R.; Caroleo, M.C.; Plastina, P.; Gallelli, L. Quercetin, Epigallocatechin Gallate, Curcumin, and Resveratrol: From Dietary Sources to Human MicroRNA Modulation. Molecules 2019, 25, 63.
- Vallino, L.; Ferraresi, A.; Vidoni, C.; Secomandi, E.; Esposito, A.; Dhanasekaran, D.N.; Isidoro, C. Modulation of non-coding RNAs by resveratrol in ovarian cancer cells: In silico analysis and literature review of the anti-cancer pathways involved. J. Tradit. Complement. Med. 2020, 10, 217–229.
- Hatami, M.; Abdolahi, M.; Soveyd, N.; Djalali, M.; Togha, M.; Honarvar, N.M. Molecular Mechanisms of Curcumin in Neuroinflammatory Disorders: A Mini Review of Current Evidences. Endocr. Metab. Immune Disord.-Drug Targets 2019, 19, 247–258.
- Kalea, A.Z.; Drosatos, K.; Buxton, J.L. Nutriepigenetics and cardiovascular disease. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 252–259.
- Pham, T.X.; Lee, J. Dietary Regulation of Histone Acetylases and Deacetylases for the Prevention of Metabolic Diseases. Nutrients 2012, 4, 1868–1886.
- Rajendran, P.; Abdelsalam, S.A.; Renu, K.; Veeraraghavan, V.; Ben Ammar, R.; Ahmed, E.A. Polyphenols as Potent Epigenetics Agents for Cancer. Int. J. Mol. Sci. 2022, 23, 11712.
- Ramos-Lopez, O.; Milagro, F.I.; Riezu-Boj, J.I.; Martinez, J.A. Epigenetic signatures underlying inflammation: An interplay of nutrition, physical activity, metabolic diseases, and environmental factors for personalized nutrition. Inflamm. Res. 2020, 70, 29–49.
- Ferraresi, A.; Phadngam, S.; Morani, F.; Galetto, A.; Alabiso, O.; Chiorino, G.; Isidoro, C. Resveratrol inhibits IL-6-induced ovarian cancer cell migration through epigenetic up-regulation of autophagy. Mol. Carcinog. 2017, 56, 1164–1181.
- Carlos-Reyes, Á.; Lopez-Gonzalez, J.S.; Meneses-Flores, M.; Gallardo-Rincón, D.; Ruíz-García, E.; Marchat, L.; La Vega, H.A.-D.; De La Cruz, O.N.H.; López-Camarillo, C. Dietary Compounds as Epigenetic Modulating Agents in Cancer. Front. Genet. 2019, 10, 79.
- Zam, W.; Khadour, A. Impact of Phytochemicals and Dietary Patterns on Epigenome and Cancer. Nutr. Cancer 2017, 69, 184–200.