Alpha-synuclein (αS) is a small, presynaptic neuronal protein encoded by the SNCA gene. Point mutations and gene multiplication of SNCA cause rare familial forms of Parkinson’s disease (PD). Misfolded αS is cytotoxic and is a component of Lewy bodies, which are a pathological hallmark of PD. Because SNCA multiplication is sufficient to cause full-blown PD, gene dosage likely has a strong impact on pathogenesis. In sporadic PD, increased SNCA expression resulting from a minor genetic background and various environmental factors may contribute to pathogenesis in a complementary manner. With respect to genetic background, several risk loci neighboring the SNCA gene have been identified, and epigenetic alterations, such as CpG methylation and regulatory histone marks, are considered important factors. These alterations synergistically upregulate αS expression and some post-translational modifications of αS facilitate its translocation to the nucleus.
- alpha-synuclein
- bioinformatics
- epigenome
- organoids
1. Introduction
2. Interaction between αS and Epigenetic Factors
2.1. Transcriptional Regulation of SNCA
2.2. Effect of αS Nuclear Localization
2.3. Interplay between Alpha-Synuclein and DNA
2.3.1. Direct Binding to DNA
2.3.2. Interaction with DNA-Modifying Proteins
2.4. Interplay between Alpha-Synuclein and Histones
2.4.1. Histone Modification via Acetylation
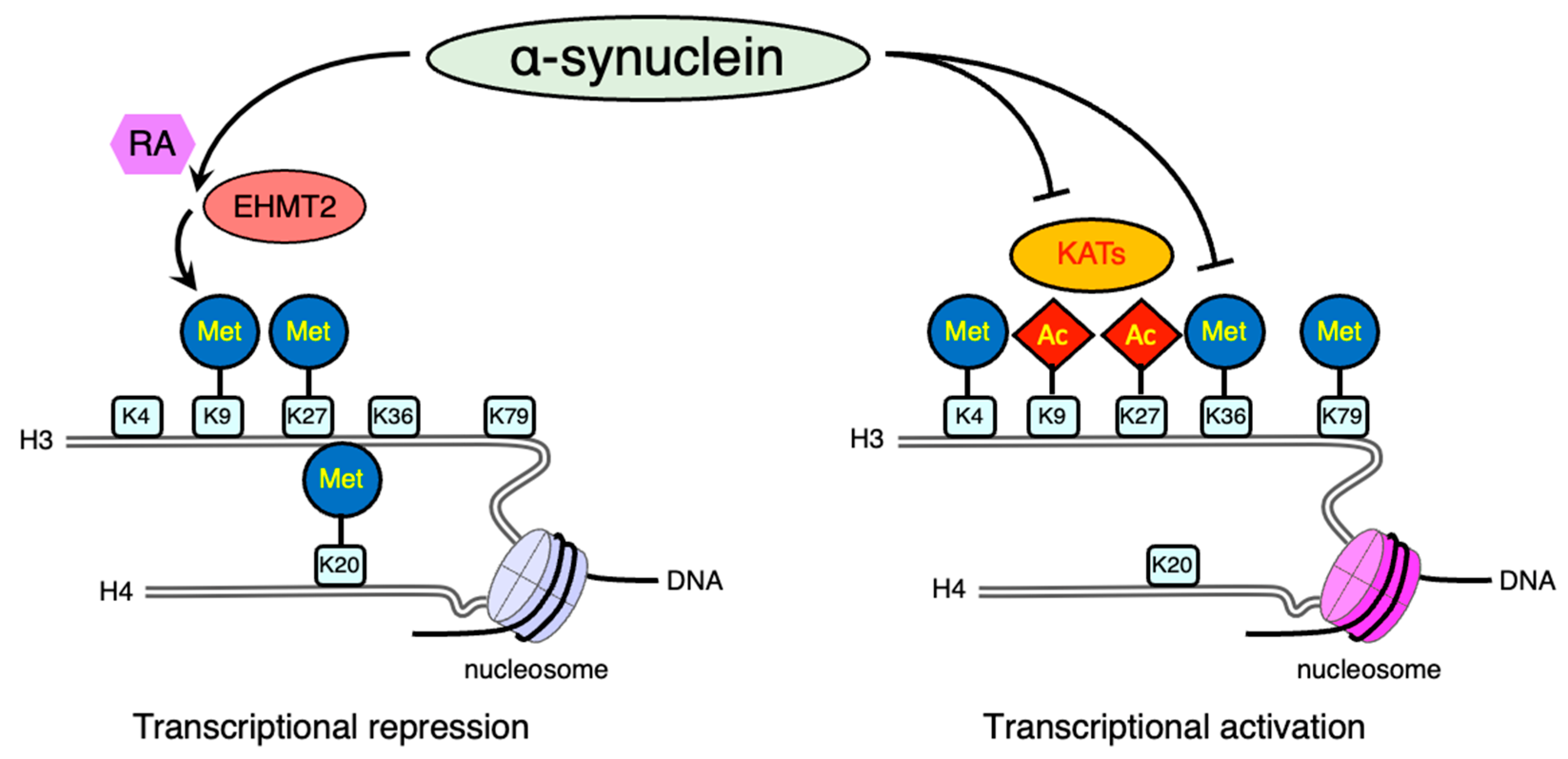
2.4.2. Histone Modification via Methylation
2.4.3. SWI/SNF Chromatin Remodeling Complexes
This entry is adapted from the peer-reviewed paper 10.3390/ijms24076645
References
- Kawahata, I.; Finkelstein, D.I.; Fukunaga, K. Pathogenic Impact of alpha-Synuclein Phosphorylation and Its Kinases in alpha-Synucleinopathies. Int. J. Mol. Sci. 2022, 23, 6216.
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815.
- Sulzer, D.; Edwards, R.H. The physiological role of alpha-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486.
- Thomas, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet. 2007, 16, R183–R194.
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006.
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841.
- Farrer, M.; Kachergus, J.; Forno, L.; Lincoln, S.; Wang, D.S.; Hulihan, M.; Maraganore, D.; Gwinn-Hardy, K.; Wszolek, Z.; Dickson, D.; et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 2004, 55, 174–179.
- Chiba-Falek, O.; Lopez, G.J.; Nussbaum, R.L. Levels of alpha-synuclein mRNA in sporadic Parkinson disease patients. Mov. Disord. 2006, 21, 1703–1708.
- Kang, J.H.; Irwin, D.J.; Chen-Plotkin, A.S.; Siderowf, A.; Caspell, C.; Coffey, C.S.; Waligorska, T.; Taylor, P.; Pan, S.; Frasier, M.; et al. Association of cerebrospinal fluid beta-amyloid 1-42, T-tau, P-tau181, and alpha-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurol. 2013, 70, 1277–1287.
- Somayaji, M.; Cataldi, S.; Choi, S.J.; Edwards, R.H.; Mosharov, E.V.; Sulzer, D. A dual role for alpha-synuclein in facilitation and depression of dopamine release from substantia nigra neurons in vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 32701–32710.
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667.
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3301–3305.
- Sugeno, N.; Takeda, A.; Hasegawa, T.; Kobayashi, M.; Kikuchi, A.; Mori, F.; Wakabayashi, K.; Itoyama, Y. Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 2008, 283, 23179–23188.
- Sugeno, N.; Hasegawa, T.; Tanaka, N.; Fukuda, M.; Wakabayashi, K.; Oshima, R.; Konno, M.; Miura, E.; Kikuchi, A.; Baba, T.; et al. Lys-63-linked ubiquitination by E3 ubiquitin ligase Nedd4-1 facilitates endosomal sequestration of internalized alpha-synuclein. J. Biol. Chem. 2014, 289, 18137–18151.
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239.
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100.
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370.
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73.
- Eschbach, J.; von Einem, B.; Muller, K.; Bayer, H.; Scheffold, A.; Morrison, B.E.; Rudolph, K.L.; Thal, D.R.; Witting, A.; Weydt, P.; et al. Mutual exacerbation of peroxisome proliferator-activated receptor gamma coactivator 1alpha deregulation and alpha-synuclein oligomerization. Ann. Neurol. 2015, 77, 15–32.
- Doll, S.G.; Meshkin, H.; Bryer, A.J.; Li, F.; Ko, Y.H.; Lokareddy, R.K.; Gillilan, R.E.; Gupta, K.; Perilla, J.R.; Cingolani, G. Recognition of the TDP-43 nuclear localization signal by importin alpha1/beta. Cell Rep. 2022, 39, 111007.
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.Y.J.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M.; et al. Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 2018, 173, 693–705.e22.
- Geertsma, H.M.; Suk, T.R.; Ricke, K.M.; Horsthuis, K.; Parmasad, J.A.; Fisk, Z.A.; Callaghan, S.M.; Rousseaux, M.W.C. Constitutive nuclear accumulation of endogenous alpha-synuclein in mice causes motor impairment and cortical dysfunction, independent of protein aggregation. Hum. Mol. Genet. 2022, 31, 3613–3628.
- Nishie, M.; Mori, F.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. A quantitative investigation of neuronal cytoplasmic and intranuclear inclusions in the pontine and inferior olivary nuclei in multiple system atrophy. Neuropathol. Appl. Neurobiol. 2004, 30, 546–554.
- Weston, L.J.; Bowman, A.M.; Osterberg, V.R.; Meshul, C.K.; Woltjer, R.L.; Unni, V.K. Aggregated Alpha-Synuclein Inclusions within the Nucleus Predict Impending Neuronal Cell Death in a Mouse Model of Parkinsonism. Int. J. Mol. Sci. 2022, 23, 15294.
- Timney, B.L.; Raveh, B.; Mironska, R.; Trivedi, J.M.; Kim, S.J.; Russel, D.; Wente, S.R.; Sali, A.; Rout, M.P. Simple rules for passive diffusion through the nuclear pore complex. J. Cell Biol. 2016, 215, 57–76.
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471.
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023.
- Surguchov, A. Protein-DNA interaction: One step closer to understanding the mechanism of neurodegeneration. J. Neurosci. Res. 2019, 97, 391–392.
- Rousseaux, M.W.; de Haro, M.; Lasagna-Reeves, C.A.; De Maio, A.; Park, J.; Jafar-Nejad, P.; Al-Ramahi, I.; Sharma, A.; See, L.; Lu, N.; et al. TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. Elife 2016, 5, e19809.
- Chen, V.; Moncalvo, M.; Tringali, D.; Tagliafierro, L.; Shriskanda, A.; Ilich, E.; Dong, W.; Kantor, B.; Chiba-Falek, O. The mechanistic role of alpha-synuclein in the nucleus: Impaired nuclear function caused by familial Parkinson’s disease SNCA mutations. Hum. Mol. Genet. 2020, 29, 3107–3121.
- Surguchov, A. alpha-Synuclein and Mechanisms of Epigenetic Regulation. Brain. Sci. 2023, 13, 150.
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102.
- Prahl, J.D.; Pierce, S.E.; van der Schans, E.J.C.; Coetzee, G.A.; Tyson, T. The Parkinson’s disease variant rs356182 regulates neuronal differentiation independently from alpha-synuclein. Hum. Mol. Genet. 2023, 32, 1–14.
- Pihlstrom, L.; Blauwendraat, C.; Cappelletti, C.; Berge-Seidl, V.; Langmyhr, M.; Henriksen, S.P.; van de Berg, W.D.J.; Gibbs, J.R.; Cookson, M.R.; The International Parkinson Disease Genomics Consortium; et al. A comprehensive analysis of SNCA-related genetic risk in sporadic parkinson disease. Ann. Neurol. 2018, 84, 117–129.
- Chiba-Falek, O.; Nussbaum, R.L. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum. Mol. Genet. 2001, 10, 3101–3109.
- Ng, A.S.L.; Tan, Y.J.; Zhao, Y.; Saffari, S.E.; Lu, Z.; Ng, E.Y.L.; Ng, S.Y.E.; Chia, N.S.Y.; Setiawan, F.; Xu, Z.; et al. SNCA Rep1 promoter variability influences cognition in Parkinson’s disease. Mov. Disord. 2019, 34, 1232–1236.
- Pedersen, C.C.; Lange, J.; Forland, M.G.G.; Macleod, A.D.; Alves, G.; Maple-Grodem, J. A systematic review of associations between common SNCA variants and clinical heterogeneity in Parkinson’s disease. NPJ Park. Dis. 2021, 7, 54.
- Markopoulou, K.; Biernacka, J.M.; Armasu, S.M.; Anderson, K.J.; Ahlskog, J.E.; Chase, B.A.; Chung, S.J.; Cunningham, J.M.; Farrer, M.; Frigerio, R.; et al. Does alpha-synuclein have a dual and opposing effect in preclinical vs. clinical Parkinson’s disease? Park. Relat. Disord. 2014, 20, 584–589; Discussion in 584.
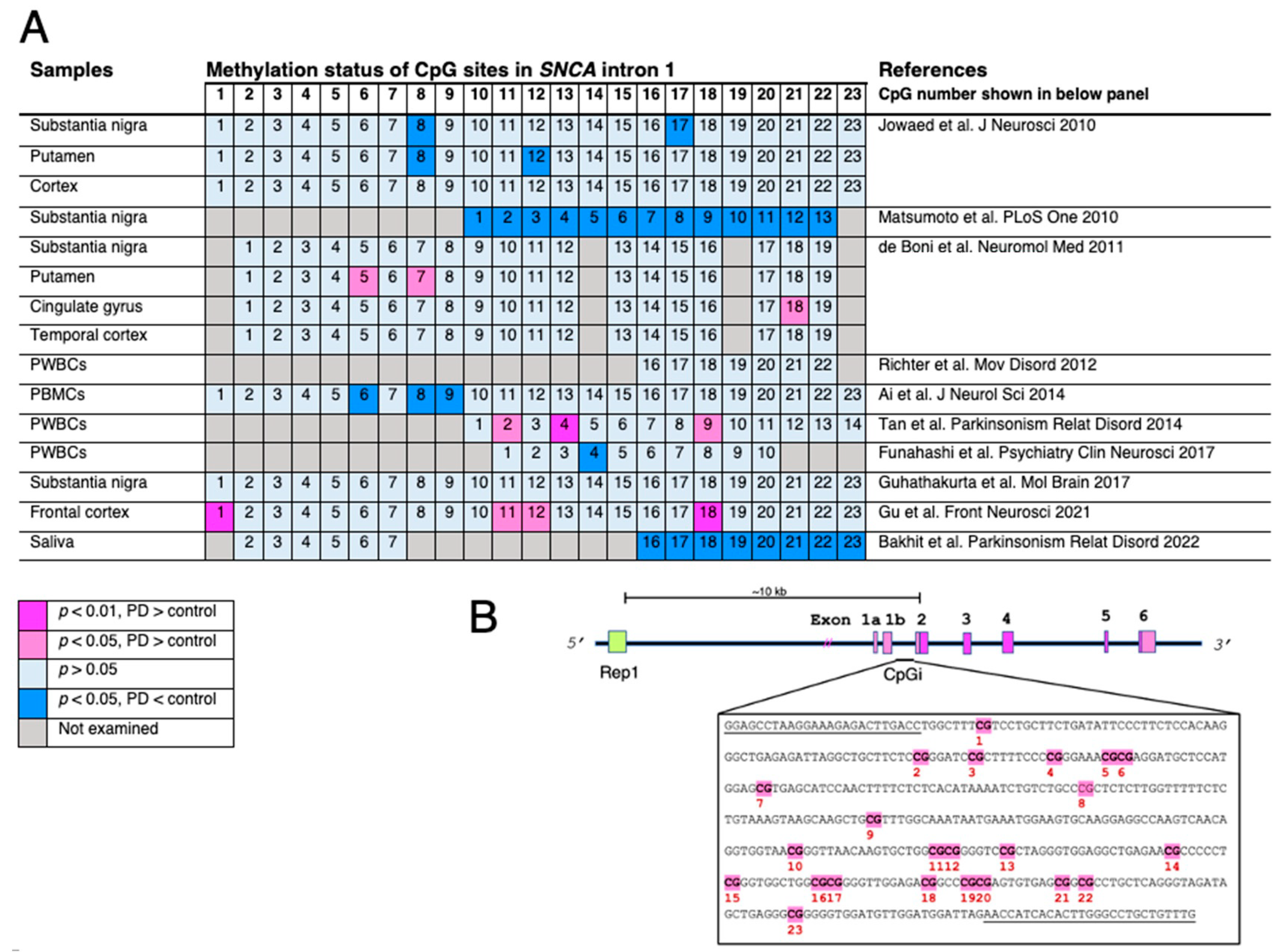
- de Boni, L.; Riedel, L.; Schmitt, I.; Kraus, T.F.J.; Kaut, O.; Piston, D.; Akbarian, S.; Wullner, U. DNA methylation levels of alpha-synuclein intron 1 in the aging brain. Neurobiol. Aging 2015, 36, 3334.e7–3334.e11.
- Miranda-Morales, E.; Meier, K.; Sandoval-Carrillo, A.; Salas-Pacheco, J.; Vazquez-Cardenas, P.; Arias-Carrion, O. Implications of DNA Methylation in Parkinson’s Disease. Front. Mol. Neurosci. 2017, 10, 225.
- Wullner, U.; Kaut, O.; deBoni, L.; Piston, D.; Schmitt, I. DNA methylation in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 108–120.
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wullner, U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359.
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 2010, 5, e15522.
- de Boni, L.; Tierling, S.; Roeber, S.; Walter, J.; Giese, A.; Kretzschmar, H.A. Next-generation sequencing reveals regional differences of the alpha-synuclein methylation state independent of Lewy body disease. Neuromol. Med. 2011, 13, 310–320.
- Richter, J.; Appenzeller, S.; Ammerpohl, O.; Deuschl, G.; Paschen, S.; Bruggemann, N.; Klein, C.; Kuhlenbaumer, G. No evidence for differential methylation of alpha-synuclein in leukocyte DNA of Parkinson’s disease patients. Mov. Disord. 2012, 27, 590–591.
- Ai, S.X.; Xu, Q.; Hu, Y.C.; Song, C.Y.; Guo, J.F.; Shen, L.; Wang, C.R.; Yu, R.L.; Yan, X.X.; Tang, B.S. Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease. J. Neurol. Sci. 2014, 337, 123–128.
- Tan, Y.Y.; Wu, L.; Zhao, Z.B.; Wang, Y.; Xiao, Q.; Liu, J.; Wang, G.; Ma, J.F.; Chen, S.D. Methylation of alpha-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Park. Relat. Disord. 2014, 20, 308–313.
- Funahashi, Y.; Yoshino, Y.; Yamazaki, K.; Mori, Y.; Mori, T.; Ozaki, Y.; Sao, T.; Ochi, S.; Iga, J.I.; Ueno, S.I. DNA methylation changes at SNCA intron 1 in patients with dementia with Lewy bodies. Psychiatry Clin. Neurosci. 2017, 71, 28–35.
- Guhathakurta, S.; Evangelista, B.A.; Ghosh, S.; Basu, S.; Kim, Y.S. Hypomethylation of intron1 of alpha-synuclein gene does not correlate with Parkinson’s disease. Mol. Brain 2017, 10, 6.
- Gu, J.; Barrera, J.; Yun, Y.; Murphy, S.K.; Beach, T.G.; Woltjer, R.L.; Serrano, G.E.; Kantor, B.; Chiba-Falek, O. Cell-Type Specific Changes in DNA Methylation of SNCA Intron 1 in Synucleinopathy Brains. Front. Neurosci. 2021, 15, 652226.
- Bakhit, Y.; Schmitt, I.; Hamed, A.; Ibrahim, E.A.A.; Mohamed, I.N.; El-Sadig, S.M.; Elseed, M.A.; Alebeed, M.A.; Shaheen, M.T.; Ibrahim, M.O.; et al. Methylation of alpha-synuclein in a Sudanese cohort. Park. Relat. Disord. 2022, 101, 6–8.
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA Expression by Targeted Editing of DNA Methylation: A Potential Strategy for Precision Therapy in PD. Mol. Ther. 2018, 26, 2638–2649.
- Guhathakurta, S.; Kim, J.; Adams, L.; Basu, S.; Song, M.K.; Adler, E.; Je, G.; Fiadeiro, M.B.; Kim, Y.S. Targeted attenuation of elevated histone marks at SNCA alleviates alpha-synuclein in Parkinson’s disease. EMBO Mol. Med. 2021, 13, e12188.
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 10919.
- Pinho, R.; Paiva, I.; Jercic, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Garcia-Esparcia, P.; Kerimoglu, C.; Pavlou, M.A.S.; Villar-Pique, A.; et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 2019, 28, 31–50.
- Weston, L.J.; Stackhouse, T.L.; Spinelli, K.J.; Boutros, S.W.; Rose, E.P.; Osterberg, V.R.; Luk, K.C.; Raber, J.; Weissman, T.A.; Unni, V.K. Genetic deletion of Polo-like kinase 2 reduces alpha-synuclein serine-129 phosphorylation in presynaptic terminals but not Lewy bodies. J. Biol. Chem. 2021, 296, 100273.
- Schell, H.; Hasegawa, T.; Neumann, M.; Kahle, P.J. Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience 2009, 160, 796–804.
- Ryu, S.; Baek, I.; Liew, H. Sumoylated alpha-synuclein translocates into the nucleus by karyopherin 6. Mol. Cell Toxicol. 2019, 15, 103–109.
- Pieger, K.; Schmitt, V.; Gauer, C.; Giessl, N.; Prots, I.; Winner, B.; Winkler, J.; Brandstatter, J.H.; Xiang, W. Translocation of Distinct Alpha Synuclein Species from the Nucleus to Neuronal Processes during Neuronal Differentiation. Biomolecules 2022, 12, 1108.
- De Giorgi, F.; Abdul-Shukkoor, M.B.; Kashyrina, M.; Largitte, L.A.; De Nuccio, F.; Kauffmann, B.; Lends, A.; Laferriere, F.; Bonhommeau, S.; Lofrumento, D.D.; et al. Neurons with Cat’s Eyes: A Synthetic Strain of alpha-Synuclein Fibrils Seeding Neuronal Intranuclear Inclusions. Biomolecules 2022, 12, 436.
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of alpha-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol. Neurodegener. 2018, 13, 21.
- Vasudevaraju, P.; Guerrero, E.; Hegde, M.L.; Collen, T.B.; Britton, G.B.; Rao, K.S. New evidence on alpha-synuclein and Tau binding to conformation and sequence specific GC* rich DNA: Relevance to neurological disorders. J. Pharm. Bioallied Sci. 2012, 4, 112–117.
- Hegde, M.L.; Vasudevaraju, P.; Rao, K.J. DNA induced folding/fibrillation of alpha-synuclein: New insights in Parkinson’s disease. Front. Biosci. Landmark Ed. 2010, 15, 418–436.
- Dent, S.E.; King, D.P.; Osterberg, V.R.; Adams, E.K.; Mackiewicz, M.R.; Weissman, T.A.; Unni, V.K. Phosphorylation of the aggregate-forming protein alpha-synuclein on serine-129 inhibits its DNA-bending properties. J. Biol. Chem. 2022, 298, 101552.
- Siddiqui, A.; Chinta, S.J.; Mallajosyula, J.K.; Rajagopolan, S.; Hanson, I.; Rane, A.; Melov, S.; Andersen, J.K. Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: Implications for Parkinson’s disease. Free Radic. Biol. Med. 2012, 53, 993–1003.
- Desplats, P.; Spencer, B.; Crews, L.; Pathel, P.; Morvinski-Friedmann, D.; Kosberg, K.; Roberts, S.; Patrick, C.; Winner, B.; Winkler, J.; et al. alpha-Synuclein induces alterations in adult neurogenesis in Parkinson disease models via p53-mediated repression of Notch1. J. Biol. Chem. 2012, 287, 31691–31702.
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92.
- Kaur, G.; Rathod, S.S.S.; Ghoneim, M.M.; Alshehri, S.; Ahmad, J.; Mishra, A.; Alhakamy, N.A. DNA Methylation: A Promising Approach in Management of Alzheimer’s Disease and Other Neurodegenerative Disorders. Biology 2022, 11, 90.
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534.
- Li, D.; Liang, J.; Guo, W.; Zhang, Y.; Wu, X.; Zhang, W. Integrative analysis of DNA methylation and gene expression data for the diagnosis and underlying mechanism of Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 971528.
- Masliah, E.; Dumaop, W.; Galasko, D.; Desplats, P. Distinctive patterns of DNA methylation associated with Parkinson disease: Identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 2013, 8, 1030–1038.
- Somayaji, M.; Lanseur, Z.; Choi, S.J.; Sulzer, D.; Mosharov, E.V. Roles for alpha-Synuclein in Gene Expression. Genes 2021, 12, 1166.
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037.
- Schaffner, S.L.; Wassouf, Z.; Lazaro, D.F.; Xylaki, M.; Gladish, N.; Lin, D.T.S.; MacIsaac, J.; Ramadori, K.; Hentrich, T.; Schulze-Hentrich, J.M.; et al. Alpha-synuclein overexpression induces epigenomic dysregulation of glutamate signaling and locomotor pathways. Hum. Mol. Genet. 2022, 31, 3694–3714.
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012, 13, 436–447.
- Park, J.; Lee, K.; Kim, K.; Yi, S.J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal Transduct. Target. Ther. 2022, 7, 217.
- Nitsch, S.; Zorro Shahidian, L.; Schneider, R. Histone acylations and chromatin dynamics: Concepts, challenges, and links to metabolism. EMBO Rep. 2021, 22, e52774.
- Farrelly, L.A.; Thompson, R.E.; Zhao, S.; Lepack, A.E.; Lyu, Y.; Bhanu, N.V.; Zhang, B.; Loh, Y.E.; Ramakrishnan, A.; Vadodaria, K.C.; et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 2019, 567, 535–539.
- Lepack, A.E.; Werner, C.T.; Stewart, A.F.; Fulton, S.L.; Zhong, P.; Farrelly, L.A.; Smith, A.C.W.; Ramakrishnan, A.; Lyu, Y.; Bastle, R.M.; et al. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 2020, 368, 197–201.
- Jos, S.; Gogoi, H.; Prasad, T.K.; Hurakadli, M.A.; Kamariah, N.; Padmanabhan, B.; Padavattan, S. Molecular insights into alpha-synuclein interaction with individual human core histones, linker histone, and dsDNA. Protein Sci. 2021, 30, 2121–2131.
- Jiang, P.; Gan, M.; Yen, S.H.; McLean, P.J.; Dickson, D.W. Histones facilitate alpha-synuclein aggregation during neuronal apoptosis. Acta Neuropathol. 2017, 133, 547–558.
- Li, P.; Ge, J.; Li, H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 96–115.
- Mazzocchi, M.; Wyatt, S.L.; Mercatelli, D.; Morari, M.; Morales-Prieto, N.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. Gene Co-expression Analysis Identifies Histone Deacetylase 5 and 9 Expression in Midbrain Dopamine Neurons and as Regulators of Neurite Growth via Bone Morphogenetic Protein Signaling. Front. Cell. Dev. Biol. 2019, 7, 191.
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537.
- Mazzocchi, M.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. The class II histone deacetylases as therapeutic targets for Parkinson’s disease. Neuronal Signal. 2020, 4, NS20200001.
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349.
- Chawla, S.; Vanhoutte, P.; Arnold, F.J.; Huang, C.L.; Bading, H. Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J. Neurochem. 2003, 85, 151–159.
- Kutil, Z.; Meleshin, M.; Baranova, P.; Havlinova, B.; Schutkowski, M.; Barinka, C. Characterization of the class IIa histone deacetylases substrate specificity. FASEB J. 2022, 36, e22287.
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Yang, Y.; Anantharam, V.; Kanthasamy, A.G. alpha-Synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J. Neurosci. 2011, 31, 2035–2051.
- Lee, J.Y.; Kim, H.; Jo, A.; Khang, R.; Park, C.H.; Park, S.J.; Kwag, E.; Shin, J.H. alpha-Synuclein A53T Binds to Transcriptional Adapter 2-Alpha and Blocks Histone H3 Acetylation. Int. J. Mol. Sci. 2021, 22, 5392.
- Renani, P.G.; Taheri, F.; Rostami, D.; Farahani, N.; Abdolkarimi, H.; Abdollahi, E.; Taghizadeh, E.; Gheibi Hayat, S.M. Involvement of aberrant regulation of epigenetic mechanisms in the pathogenesis of Parkinson’s disease and epigenetic-based therapies. J. Cell Physiol. 2019, 234, 19307–19319.
- Wu, Q.; Yang, X.; Zhang, L.; Zhang, Y.; Feng, L. Nuclear Accumulation of Histone Deacetylase 4 (HDAC4) Exerts Neurotoxicity in Models of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 6970–6983.
- Paiva, I.; Pinho, R.; Pavlou, M.A.; Hennion, M.; Wales, P.; Schutz, A.L.; Rajput, A.; Szego, E.M.; Kerimoglu, C.; Gerhardt, E.; et al. Sodium butyrate rescues dopaminergic cells from alpha-synuclein-induced transcriptional deregulation and DNA damage. Hum. Mol. Genet. 2017, 26, 2231–2246.
- Zhang, Y.; Xu, S.; Qian, Y.; He, X.; Mo, C.; Yang, X.; Xiao, Q. Sodium butyrate attenuates rotenone-induced toxicity by activation of autophagy through epigenetically regulating PGC-1alpha expression in PC12 cells. Brain Res. 2022, 1776, 147749.
- Mazzocchi, M.; Goulding, S.R.; Wyatt, S.L.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. LMK235, a small molecule inhibitor of HDAC4/5, protects dopaminergic neurons against neurotoxin- and alpha-synuclein-induced degeneration in cellular models of Parkinson’s disease. Mol. Cell. Neurosci. 2021, 115, 103642.
- Harrison, I.F.; Powell, N.M.; Dexter, D.T. The histone deacetylase inhibitor nicotinamide exacerbates neurodegeneration in the lactacystin rat model of Parkinson’s disease. J. Neurochem. 2019, 148, 136–156.
- Hasegawa, T.; Baba, T.; Kobayashi, M.; Konno, M.; Sugeno, N.; Kikuchi, A.; Itoyama, Y.; Takeda, A. Role of TPPP/p25 on alpha-synuclein-mediated oligodendroglial degeneration and the protective effect of SIRT2 inhibition in a cellular model of multiple system atrophy. Neurochem. Int. 2010, 57, 857–866.
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519.
- Liu, Y.; Zhang, Y.; Zhu, K.; Chi, S.; Wang, C.; Xie, A. Emerging Role of Sirtuin 2 in Parkinson’s Disease. Front. Aging Neurosci. 2019, 11, 372.
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.L.; Chua, K.; et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 2008, 28, 1688–1701.
- Di Nisio, E.; Lupo, G.; Licursi, V.; Negri, R. The Role of Histone Lysine Methylation in the Response of Mammalian Cells to Ionizing Radiation. Front. Genet. 2021, 12, 639602.
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24.
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 arginine methyltransferase: Many roles in development, cancer and beyond. Cell. Mol. Life Sci. 2015, 72, 2041–2059.
- Chen, K.; Bennett, S.A.; Rana, N.; Yousuf, H.; Said, M.; Taaseen, S.; Mendo, N.; Meltser, S.M.; Torrente, M.P. Neurodegenerative Disease Proteinopathies Are Connected to Distinct Histone Post-translational Modification Landscapes. ACS Chem. Neurosci. 2018, 9, 838–848.
- Sugeno, N.; Jackel, S.; Voigt, A.; Wassouf, Z.; Schulze-Hentrich, J.; Kahle, P.J. alpha-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci. Rep. 2016, 6, 36328.
- Garcia-Reitbock, P.; Anichtchik, O.; Bellucci, A.; Iovino, M.; Ballini, C.; Fineberg, E.; Ghetti, B.; Della Corte, L.; Spano, P.; Tofaris, G.K.; et al. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 2010, 133 Pt 7, 2032–2044.
- Cenik, B.K.; Shilatifard, A. COMPASS and SWI/SNF complexes in development and disease. Nat. Rev. Genet. 2021, 22, 38–58.
- Ho, L.; Jothi, R.; Ronan, J.L.; Cui, K.; Zhao, K.; Crabtree, G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc. Natl. Acad. Sci. USA 2009, 106, 5187–5191.
- Sun, L.; Zhang, J.; Chen, W.; Chen, Y.; Zhang, X.; Yang, M.; Xiao, M.; Ma, F.; Yao, Y.; Ye, M.; et al. Attenuation of epigenetic regulator SMARCA4 and ERK-ETS signaling suppresses aging-related dopaminergic degeneration. Aging Cell 2020, 19, e13210.
- Sanphui, P.; Kumar Das, A.; Biswas, S.C. Forkhead Box O3a requires BAF57, a subunit of chromatin remodeler SWI/SNF complex for induction of p53 up-regulated modulator of apoptosis (Puma) in a model of Parkinson’s disease. J. Neurochem. 2020, 154, 547–561.