α-Synuclein (αS) is a relatively small protein with a molecular weight of 14.5 kDa. It consists of 140 amino acids encoded by the
SNCA gene on chromosome 4q22.1 [
1]. The first synuclein cDNA was isolated from the electric organ of
Torpedo californica and was primarily detected in the nuclear envelope and presynaptic nerve terminals [
2]. Thus, the name αS is derived from a combination of the prefixes for synapse (“syn”), nucleus (“nucl”), and the suffix for protein (“ein”). αS is considered an important molecule that triggers the neurodegenerative process in synucleinopathies, including Parkinson’s disease (PD) [
3]. Most cases of PD are sporadic; however, less than 10% have a family history and several genes associated with the inherited forms of PD have been identified [
4].
SNCA was the first familial PD gene (PARK1) discovered, and patients harboring missense mutations exhibit classic adult-onset forms of PD [
5].
SNCA can cause PD not only through a point mutation, but also by gene multiplication, with the latter designated as
PARK4 [
6,
7]. These copy number changes correlate with elevated transcript levels of
SNCA and subsequent increase of αS protein production [
6]. In addition, increased
SNCA mRNA was observed predominantly in the midbrain or substantia nigra of patients with PD [
8], while a decrease is detected in other tissues and regions, such as temporal and frontal cortexes and cerebrospinal fluid [
9]. It is assumed that this tissue-specific increase in
SNCA is closely related to neurodegeneration. Thereby, the idea that increased αS expression in the nervous system can cause dopaminergic neuron loss is the rationale for using αS overexpressing cell and animal models for PD studies.
2. Interaction between αS and Epigenetic Factors
2.1. Transcriptional Regulation of SNCA
Comprehensive genome-wide association studies (GWASs) have examined the association between SNPs and the development of idiopathic PD. Several risk loci in the
SNCA gene have been identified [
32]. A meta-analysis revealed that rs356182 is the most significant SNPs associated with PD [
32]; however, it is debatable whether the rs356168 risk variant acts directly on
SNCA regulation or is involved in a neurodegenerative process unrelated to αS function [
33,
34]. In addition, the complex polymorphic microsatellite repeat site Rep1, located approximately 10 kb upstream of the
SNCA transcription start site, has been reported [
35]. Longer Rep1 alleles increase
SNCA gene expression compared with shorter alleles [
35]; however, the effect of the Rep allele on disease severity and the risk of cognitive decline remains controversial [
36,
37,
38].
In addition to gene mutations, epigenetic regulation is important to the regulation of
SNCA expression. DNA methylation occurs on cytosine residues located adjacent to guanine, which are known as CpG sites. The CpG site near the transcription start site, known as the CpG island, and its methylation, represses the transcription of the associated gene. The
SNCA CpG island is located in intron 1, which is upstream of the initiation codon [
39]. It serves as a binding site for several transcription factors that regulate
SNCA expression [
40] (
Figure 1). Hypomethylation of this region results in increased expression of
SNCA, which leads to the accumulation of αS and ultimately, neurotoxicity [
41]. In a study of post-mortem brains, a marked reduction in
SNCA methylation was observed in the substantia nigra, putamen, and cortex of PD patients [
42]. Although PD-related alterations in methylation status in intron 1 is controversial [
43,
44,
45,
46,
47,
48,
49,
50,
51], the induction of robust hypermethylation of
SNCA CpG island results in a reduction in
SNCA expression, which may be exploited as a therapeutic approach to prevent pathogenic αS accumulation [
52]. This intron 1 domain is also associated with histone modification, and another epigenetic modulator, H3K4me3 transcriptional active mark, was more prevalent in the substantia nigra of PD brains [
53]. Based on these observations, attempts have been made to regulate epigenetically active histone marks by gene editing in an experimental model of PD. Precisely, a deletion of H3K4me3 by locus-specific editing successfully reduced αS in SH-SY5Y neuronal cells and iPSC-derived dopaminergic neurons [
53].
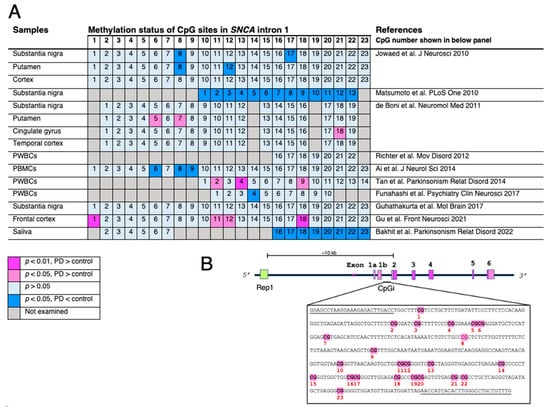
Figure 1. Changes in methylation status of CpG islands located in
SNCA intron 1. (
A) The indexed number of CpG sites is based on the report by Jowaed et al. [
42,
43,
44,
45,
46,
47,
48,
49,
50,
51]. The numbering of CpG sites is not identical in each study and is, therefore, corrected and displayed accordingly. Differences in methylation rates of CpG sites are compared between Parkinson’s disease (PD) or dementia with Lewy bodies (DLB) and healthy subjects or disease controls. The analyses are varied, but do not show consistent results. Nevertheless, CpG numbers 8 and 18 are likely more sensitive to the methylation modulating system or nuclear environment. (
B) Schematic presentation of the human
SNCA gene. Rep1, a dinucleotide repeat site, the length of which affects
SNCA expression, located approximately 10 kb upstream from the start codon in exon 2. The CpG island located in intron 1 includes 23 CpG sites. Primer sequences for pyrosequencing by Jowaed et al. are underlined. The abbreviations used are as follows: PWBCs, peripheral white blood cells; PBMC, peripheral blood mononuclear cells.
2.2. Effect of αS Nuclear Localization
As noted above, αS does not have a canonical NLS; but, post-translational modifications and other factors affect its nuclear localization. In neuronal inclusion bodies (i.e., Lewy bodies) of PD brains, greater than 90% of the αS is highly phosphorylated at serine 129 (S129), which is used as a marker for pathological diagnosis. In vivo experiments using mouse brain and primary cortical neurons have revealed that S129 phosphorylated αS is rapidly translocated to the nucleus of laser-induced focal lesions [
54]. Similarly, in H4 human neuroglioma cells, S129 phosphorylated αS exhibited higher affinity toward the nucleus; whereas, downregulated cyclin B1 and E2F transcription factor 8 genes were involved in the cell cycle [
55]. Polo-like kinase 2 (PLK2), an enzyme that catalyzes the S129 phosphorylation of αS, may be involved in the nuclear-cytoplasmic shuttling of αS [
56]. The presence of S129 phospho-αS may be associated with aging, because S129 phosphorylated αS was observed in the nucleus of aged mice, but not in young A30P human αS transgenic mice [
57].
Besides phosphorylation, SUMOylation, which is generated by mature small ubiquitin-related modifiers (SUMO), may occur during the nuclear translocation of αS [
58]. Interestingly, distinct αS species translocate from the nucleus to neuronal processes during neuronal differentiation, which suggests that the maturation process of the nervous system may affect subcellular localization [
59]. With respect to protein conformational change and aggregation, exposure to human fibrotic αS seed facilitates the formation of intranuclear inclusions in mouse primary cortical neurons [
60]. Furthermore, the inoculation of αS preformed fibrils into the stomach wall of wild-type mice resulted in the formation of a small number of nuclear inclusions in the dorsal motor nucleus of the vagal nerve [
61]. These results suggest that the progression of PD may contribute to the aberrant accumulation of misfolded αS in neuronal nuclei.
2.3. Interplay between Alpha-Synuclein and DNA
2.3.1. Direct Binding to DNA
αS can bind to supercoiled DNA in a conformation-specific manner and alter the bending properties and stability of DNA, which in turn modulates the conformation status of αS itself [
62,
63,
64]. αS also directly binds to a large subset of DNA promoter sequences, including the
PPARGC1A (PGC-1α) and
NOTCH1 gene promoters, thereby downregulating the transcription of their target genes [
55,
65,
66]. Interestingly, αS colocalizes with DNA damage response components to form discrete foci in the neuronal nuclei and the removal of αS decreases the repair of DNA double-strand breaks [
54], suggesting a neuroprotective function of endogenous αS.
2.3.2. Interaction with DNA-Modifying Proteins
DNA methylation, which is an important epigenetic process, is catalyzed by multiple DNA methyltransferases (DNMTs: DNMT1, DNMT2, DNMT3, DNMT3L) through the transfer of a methyl group from
S-adenyl methionine (SAM) to the fifth carbon of cytosine, resulting in 5mC [
67]. During DNA replication, DNMT1 retrieves the DNA methylation prototype from the parent DNA strand and transfers it to the newly synthesized daughter strand [
68]. Conversely, DNA demethylation is often catalyzed by enzymes of the ten-eleven translocation (TET) family, which counteract the activity of DNMTs [
69]. In recent years, DNA methylation has been a significant area of interest in PD research [
70]. Given that CpG methylation profiles associated with PD matched approximately 30% between brain tissue and blood samples [
71], specific loci could be identified as candidate biomarkers for PD in peripheral blood mononuclear cells [
72]. An early study demonstrated that DNMT1, normally located in the nucleus, was sequestered in the cytoplasm following αS overexpression, causing global DNA hypomethylation and transcriptional activation of downstream genes [
73]. A subsequent study also found that altered DNA methylation at CpG sites affected gene expression associated with locomotor behavior and the glutamate signaling pathway [
74].
2.4. Interplay between Alpha-Synuclein and Histones
A nucleosome is a fundamental unit of chromatin consisting of 147 base pairs of DNA and an octamer of core histone proteins containing two copies of each of the histones: H2A, H2B, H3, and H4 [
75]. The amino acid sequences of the histone proteins are conserved across species from
Archaeum to
Homo sapiens [
76]. The chromosome structure of eukaryotic cells may be divided into two regions: heterochromatin, which is tightly condensed and transcriptionally repressed, and euchromatin, which is untangled and transcriptionally active [
76]. The condensation of histones is essentially responsible for the organization of euchromatin and heterochromatin. The chromatin status is defined by the types of post-translational modifications in histone tails, including acetylation, dopaminylation, methylation, phosphorylation, serotonylation, SUMOylation, and ubiquitination [
77,
78,
79]. These histone modifications may alter the affinity to DNA and other histones by altering the surface charge; thereby, reversibly regulating the entry of transcription factors and other transcription-related proteins [
76].
2.4.1. Histone Modification via Acetylation
αS binds to the N-terminal flexible tails of histones H3, H4, and H1 [
80]. Its fibrillation is accelerated by H1 released from the nucleus during apoptosis [
81]. Lysine acetylation is a reversible process, which is post-translational modification that alters the charge of lysine residues and modifies protein structure to influence protein function [
82]. Histone acetylation modulates fundamental cell processes, such as transcriptional regulation and chromatin remodeling. The balance between the activities of lysine acetyltransferases (KAT) and histone deacetylases (HDAC) is strictly controlled, but can be disrupted in neurodegenerative diseases, such as PD [
83].
KAT catalyzes the transfer of an acetyl group from acetyl-CoA to the
ε-amino group of an internal lysine residue [
84]. Acetylated histones counteract the positive charge on the residue, which prevents DNA-histone interactions and activates transcription, resulting in a loose chromatin state that facilitates transcription. In contrast, histone deacetylation results in a tighter chromatin structure, which suppresses transcriptional activity [
84]. Mammalian KATs are classified into two groups according to their cellular localization: Nuclear KATs (type A) and cytoplasmic KATs (type B). Type A KATs are primarily involved in transcriptional regulation and may be further classified into five families: Gcn5-related
N-acetyltransferase (GNAT), p300/cAMP response element binding (CREB) binding protein (CBP), MYST basal transcription factors, and the nuclear receptor coactivator (NCoA) family [
82]. The reverse reaction, deacetylation, is catalyzed by histone deacetylases. To date, 18 mammalian HDACs have been identified and classified into four classes (class I, II, III, and IV) based on their sequence similarity to yeast HDACs. Class I HDACs include HDAC1, 2, 3, and 8; Class II HDACs are subdivided into class IIa (HDAC4, 5, 7, and 9) and IIb (HDAC6 and 10); Class III HDACs are members of the sirtuin family; and class IV HDAC includes only HDAC11 [
85]. Of note, nucleosome proteins are not a specific substrate for these KATs and HDACs [
86]. For example, class IIa HDACs (HDAC4, HDAC5, and HDAC7) shuttle between the nucleus and the cytoplasm [
87]. HDAC4 recognizes a variety of extra-nuclear proteins as substrates, including forkhead transcription factors of the O class (FOXO), myosin heavy chain isoforms (MyHC), PGC-1α, and heat shock cognate 71 kDa (Hsc70) [
88].
In general, histone acetylation is associated with gene activation; whereas, the removal of the acetyl mark induces a closed chromatin structure. Several studies suggest that αS reduces histone acetylation, which inhibits the expression of certain genes (
Figure 2). Although the intracellular function of αS in neurodegenerative processes remains unclear, nuclear-localized αS increases cytotoxicity [
27] whereas cytosolic αS is neuroprotective [
89]. Cytosolic αS reduces p300 levels and its KAT activity, resulting in a reduction of histone acetylation in dopaminergic neural cell lines [
89]. Alternatively, the A53T mutant αS modulates histone acetylation by interacting with transcriptional adapter 2-α (TADA2a), a component of the major histone acetyltransferase p300/CBP [
90].
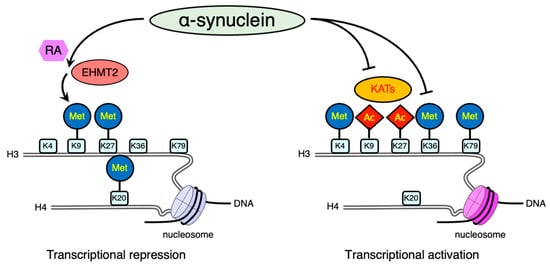
Figure 2. Schematic illustration of post-translational modifications of histone proteins affected by alpha-synuclein (αS). Lysine methylation has different functions depending on its residues. Methylation (Met) of H3 lysine 9 (K9), lysine 27 (K27), and H4 lysine 20 (K20) is often associated with transcriptional repression. In contrast, the methylation of H3 lysine 4 (K4), lysine 36 (K36), or lysine 79 (K79) is largely responsible for transcriptional activation. In addition, histone acetylation (Ac) usually promotes gene expression. Combined stimulation with αS and retinoic acid (RA) enhances K9 methylation of H3 through the activation of euchromatic histone lysine N-methyltransferase 2 (EHMT2). In addition, the inactivation of lysine acetyltransferases (KATs) decreases histone acetylation. The other transcriptional active mark, K36 methylation of H3, is also downregulated by αS. Both increased repressive signals and disruption of active marks result in the transcriptional repression of downstream genes.
Class IIa HDACs have NLS and shuttle between the nucleus and cytoplasm [
91]. HDAC4, a member of the class IIa HDACs, is abundantly expressed in neurons and accumulates in the nucleus following stimulation with MPTP (1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridines), a dopaminergic neurotoxin. Nuclear HDAC4 also mediates cell death in A53T mutant αS-expressing cells by inhibiting CREB and myocyte enhancer factor 2A (MEF2A) [
92]. The global HDAC inhibitors, sodium butyrate (NaB) and suberoylanilide hydroxamic acid (SAHA), protect against αS-mediated neurotoxicity in cell and transgenic
Drosophila models [
27]. Decreased H3 acetylation and altered RNA-seq gene expression profiles through αS in LUHMES (Lund human mesencephalic) dopaminergic cells were attenuated by adding NaB, which may be mediated by DNA repair [
93]. In contrast, the neuroprotective effects of NaB in PC12 cells are dependent on the activation of PGC-1α via hyperacetylation of its promoter region [
94]. Class I HDAC-specific inhibitors and class IIb HDAC6 inhibitors failed to alleviate the αS-induced neurite outgrowth defects in SH-SY5Y cells; whereas, the class IIa HDAC4/5 inhibitor LMK235 successfully promoted neurite outgrowth [
95]. The sirtuin family of NAD(+)-dependent class III HDACs are also candidate therapeutic targets in PD [
96]. Blockade of sirtuin 2 resulted in a dose-dependent protective effect against αS-induced toxicity [
97,
98], suggesting the potential therapeutic applications of targeting specific HDACs. The neuroprotective effects of HDAC inhibitors are currently under preclinical investigation as disease-modifying therapy for PD [
99]; however, some of their effects may be mediated by mechanisms unrelated to histone acetylation, such as microtubule stabilization [
100].
2.4.2. Histone Modification via Methylation
Lysine residues can accept mono-, di-, and tri-methylation (me1, me2, and me3) modifications; whereas, arginine residues accept asymmetric or symmetric di-methylation or mono-methylation. Although histone acetylation usually promotes gene expression, the function of histone methylation depends on the context. Methylation of histone H3 on lysine 4 (H3K4), lysine 36 (H3K36), or lysine 79 (H3K79) is largely responsible for transcriptional activation. In contrast, methylation of histone H3 at lysine 9 (H3K9) and lysine 27 (H3K27) or histone H4 on lysine 20 (H4K20) is often associated with transcriptional repression [
101]. Lysine methylation is catalyzed by lysine methyltransferases (KMTs), known as “Writers,”; whereas, histone demethylation is catalyzed by lysine demethylases (KDMs), known as “Erasers”.
Protein arginine methyltransferases (PRMTs) transfer methyl groups from
S-Adenosylmethionine (SAM) to arginine residues on histone proteins. They are classified into three types based on their catalytic activity [
102]. Type I PRMTs (PRMT1, 2, 3, 4, 6, and 8) asymmetrically di-methylate arginine residues (ADMA) whereas type II PRMTs (PRMT5 and PRMT9) symmetrically di-methylate arginine residues (SDMA). Type III (PRMT7) catalyzes only mono-methylated arginine formation (MMA) [
102]. The major targets of arginine methylation in histone proteins are histone H3 on arginine 2 (H3R2), arginine 8 (H3R8), and histone H4 on arginine 3 (H4R3). The histone code of arginine methylation is rather complicated. Both ADMA and SDMA are di-methylated, but the asymmetric type, ADMA, is often associated with transcriptional activation, whereas the symmetric type, SDMA, results in transcriptional repression [
103]. Therefore, the structural behavior of chromatin differs depending on the type of PRMT acting on the target histone tails. Consequently, SDMA occupying regional histones corresponds to the signature of type II PRMTs.
αS has been found to interact with several epigenetic writers. In an αS yeast model, the altered histone marks including H3K36 di-methylation were distinct from histone marks affected by TDP-43 or FUS [
104]. Additionally, overexpression of αS in transgenic
Drosophila and SH-SY5Y cells resulted in H3K9 di-methylation through upregulation of euchromatic histone lysine
N-methyltransferase 2 (
EHMT2) in the presence of retinoic acid [
105]. The chromatin immunoprecipitation with antibodies against repressor element-1 (RE1)-silencing transcription factor (REST) inactivated transcription, one of the EHMT2 interacting protein, revealed the repressed downstream genes
SNAP25 and
L1CAM. SNAP25 is a major component of SNARE complex involved in synaptic function; thereby, these changes may contribute to the synaptic dysfunction that occurs in PD brain [
106].
2.4.3. SWI/SNF Chromatin Remodeling Complexes
The SWI/SNF family was first functionally characterized in
Saccharomyces cerevisiae but is conserved across species throughout eukaryotes. The SWI/SNF family contains an ATPase subunit that utilizes ATP-dependent chromatin remodeling to enhance DNA accessibility during transcription [
107]. BAF (Brg1-associated factors) is a mammalian homolog of SWI/SNF, whose central ATPase is composed of SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 4 (SMARCA4), and SMARCA2. The BAF complexes are not just chromatin remodeling factors, but they can repress or activate gene expression [
108]. Interestingly, SMARCA4 harbors SNPs and a meta-analysis yielded
p-values of 1 × 10
−4 and 0.05, which may be considered a potential risk for PD, although not in the top 10,000 most significant GWAS results [
32,
109]. Furthermore, in silico disease-associated gene prediction followed by in vivo
Drosophila genetic screening identified SMARCA4. Knockdown of Brahma, the
Drosophila homolog of SMARCA4, in dopaminergic neurons prolonged the lifespan of human
LRRK2 or
SNCA transgenic
Drosophila [
109]. Another component of the BAF complex, Brg-associated factor 57 (BAF57), was modulated in PC12 cells treated with the dopaminergic neurotoxin, 6-OHDA [
110].