The immune system and autophagy share a functional relationship. Both innate and adaptive immune responses involve autophagy and, depending on the disease’s origin and pathophysiology, it may have a detrimental or positive role on autoimmune disorders. As a “double-edged sword” in tumors, autophagy can either facilitate or impede tumor growth. The autophagy regulatory network that influences tumor progression and treatment resistance is dependent on cell and tissue types and tumor stages. Several autophagy modifiers have demonstrated beneficial effects in models of autoimmune disease, emphasizing their therapeutic potential as treatments for autoimmune disorders.
- autophagy
- autoimmunity
- carcinogenesis
- immune system
- autoimmune disorders
1. Impact of the Microbiome on Autophagy in Relation to Anticancer Immunity and Self-Tolerance
2. Involvement of Autophagy in Escape from Central Tolerance and Evading Anticancer Immunity
3. Endocrine Influence on Autophagy, Autoimmunity and Carcinogenesis
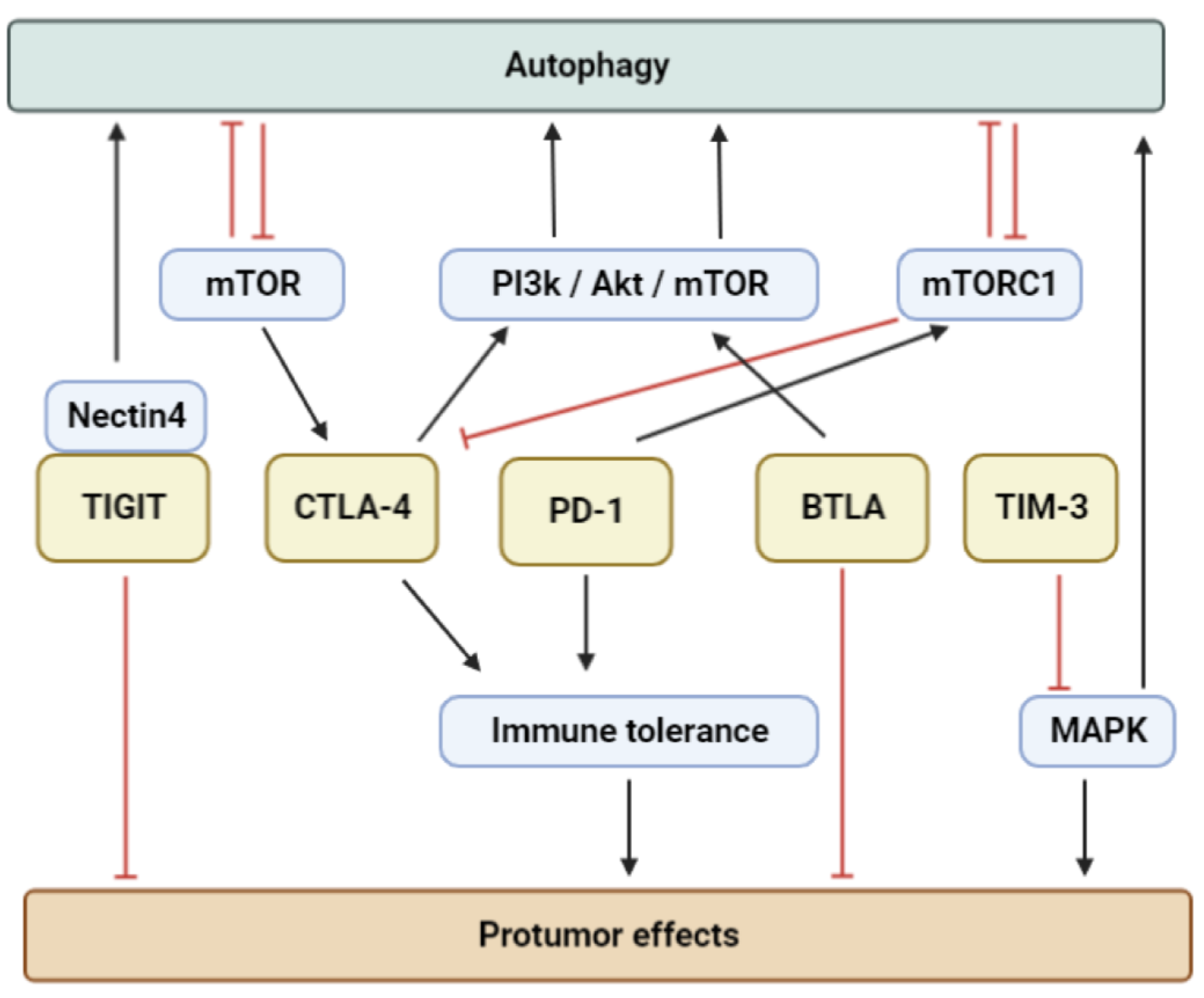
4. Immune Checkpoints, Apoptosis and Autophagy
5. Regulatory T and B Cells, Autophagy, Autoimmunity and Carcinogenesis
6. Involvement of γδT Cells in Autophagy, Autoimmunity, and Carcinogenesis
7. Myeloid-Derived Suppressor Cells and Macrophages in Relation to Autophagy, Autoimmunity and Carcinogenesis
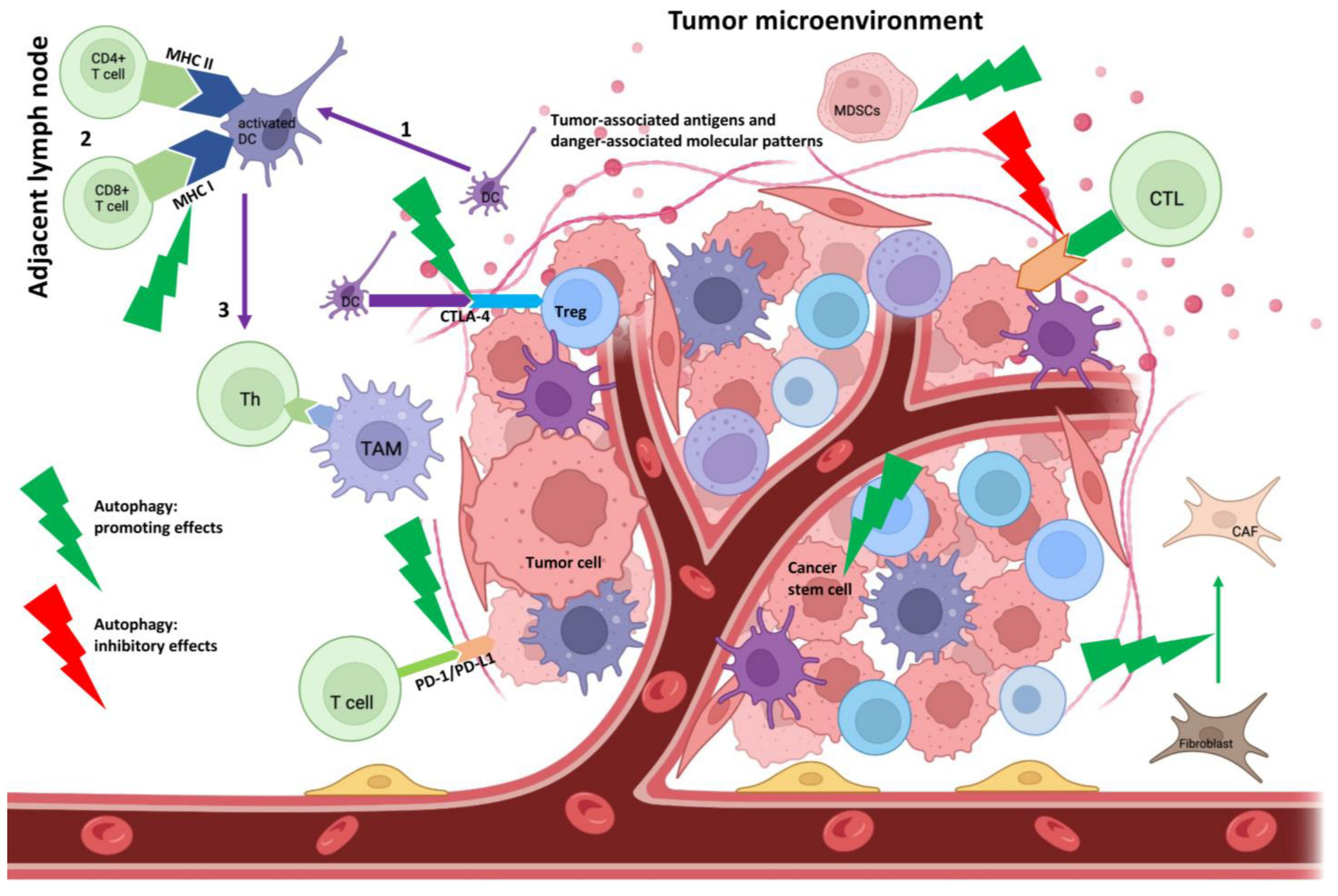
8. The Cross-Talk of Cell-to-Cell Interactions with Autophagy, Autoimmunity, and Carcinogenesis
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11041130
References
- Kaźmierczak-Siedlecka, K.; Daca, A.; Fic, M.; van de Wetering, T.; Folwarski, M.; Makarewicz, W. Therapeutic methods of gut microbiota modification in colorectal cancer management—fecal microbiota transplantation, prebiotics, probiotics, and synbiotics. Gut Microbes 2020, 11, 1518–1530.
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215.
- Mäkinen, A.; Nawaz, A.; Mäkitie, A.; Meurman, J.H. Role of Non-Albicans Candida and Candida Albicans in Oral Squamous Cell Cancer Patients. J. Oral Maxillofac. Surg. 2018, 76, 2564–2571.
- Kaźmierczak-Siedlecka, K.; Daca, A.; Roviello, G.; Catalano, M.; Połom, K. Interdisciplinary insights into the link between gut microbiome and gastric carcinogenesis-what is currently known? Gastric Cancer 2022, 25, 1–10.
- Bakhti, S.Z.; Latifi-Navid, S. Interplay and cooperation of Helicobacter pylori and gut microbiota in gastric carcinogenesis. BMC Microbiol. 2021, 21, 258.
- Matson, V.; Chervin, C.S.; Gajewski, T.F. Cancer and the Microbiome—Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology 2021, 160, 600–613.
- Li, W.; Deng, Y.; Chu, Q.; Zhang, P. Gut microbiome and cancer immunotherapy. Cancer Lett. 2019, 447, 41–47.
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97.
- Michail, S.; Bultron, G.; Depaolo, R.W. Genetic variants associated with Crohn’s disease. Appl. Clin. Genet. 2013, 6, 25–32.
- Caparrós, E.; Wiest, R.; Scharl, M.; Rogler, G.; Gutiérrez Casbas, A.; Yilmaz, B.; Wawrzyniak, M.; Francés, R. Dysbiotic microbiota interactions in Crohn’s disease. Gut Microbes 2021, 13, 1949096.
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693.
- Wagner, J.; Skinner, N.A.; Catto-Smith, A.G.; Cameron, D.J.S.; Michalski, W.P.; Visvanathan, K.; Kirkwood, C.D. TLR4, IL10RA, and NOD2 mutation in paediatric Crohn’s disease patients: An association with Mycobacterium avium subspecies paratuberculosis and TLR4 and IL10RA expression. Med. Microbiol. Immunol. 2013, 202, 267–276.
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603.
- Abdelnaby, H.; Ndiaye, N.; D’Amico, F.; Fouad, A.; Hassan, S.; Elshafey, A.; Al Hashash, W.; Faisal, M.; Alshamali, Y.; Al-Taweel, T.; et al. NOD2/CARD15 polymorphisms (P268S, IVS8+158, G908R, L1007fs, R702W) among Kuwaiti patients with Crohn’s disease: A case-control study. Saudi J. Gastroenterol. 2021, 27, 249–256.
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms. Viruses 2019, 11, 762.
- Coppieters, K.T.; Wiberg, A.; von Herrath, M.G. Viral infections and molecular mimicry in type 1 diabetes. APMIS 2012, 120, 941–949.
- Rojas, M.; Restrepo-Jiménez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123.
- Rendell, M.S. Obesity and diabetes: The final frontier. Expert Rev. Endocrinol. Metab. 2023, 18, 81–94.
- An, C.; Pipia, I.; Ruiz, A.S.; Argüelles, I.; An, M.; Wase, S.; Peng, G. The molecular link between obesity and genomic instability in cancer development. Cancer Lett. 2023, 555, 216035.
- Cao, Y.; Ren, G.; Zhang, Y.; Qin, H.; An, X.; Long, Y.; Chen, J.; Yang, L. A new way for punicalagin to alleviate insulin resistance: Regulating gut microbiota and autophagy. Food Nutr. Res. 2021, 65.
- Ceccarelli, F.; Agmon-Levin, N.; Perricone, C. Genetic Factors of Autoimmune Diseases 2017. J. Immunol. Res. 2017, 2017, 2789242.
- Noble, J.A.; Valdes, A.M. Genetics of the HLA region in the prediction of type 1 diabetes. Curr. Diab Rep. 2011, 11, 533–542.
- Kulski, J.K.; Suzuki, S.; Shiina, T. Human leukocyte antigen super-locus: Nexus of genomic supergenes, SNPs, indels, transcripts, and haplotypes. Hum. Genome Var. 2022, 9, 49.
- Das, P.; Abraham, R.; David, C. HLA transgenic mice as models of human autoimmune diseases. Rev. Immunogenetics 2000, 2, 105–114.
- Rouanne, M.; Adam, J.; Radulescu, C.; Letourneur, D.; Bredel, D.; Mouraud, S.; Goubet, A.G.; Leduc, M.; Chen, N.; Tan, T.Z.; et al. BCG therapy downregulates HLA-I on malignant cells to subvert antitumor immune responses in bladder cancer. J. Clin. Invest. 2022, 132, e145666.
- Mehra, P.; Wells, A.D. Variant to Gene Mapping to Discover New Targets for Immune Tolerance. Front. Immunol. 2021, 12, 633219.
- Musette, P.; Bouaziz, J.D. B Cell Modulation Strategies in Autoimmune Diseases: New Concepts. Front. Immunol. 2018, 9, 622.
- Anderson, M.S.; Venanzi, E.S.; Chen, Z.; Berzins, S.P.; Benoist, C.; Mathis, D. The cellular mechanism of Aire control of T cell tolerance. Immunity 2005, 23, 227–239.
- Fierabracci, A. Recent insights into the role and molecular mechanisms of the autoimmune regulator (AIRE) gene in autoimmunity. Autoimmun. Rev. 2011, 10, 137–143.
- Perniola, R. Twenty Years of AIRE. Front. Immunol. 2018, 9, 98.
- Zhu, M.L.; Bakhru, P.; Conley, B.; Nelson, J.S.; Free, M.; Martin, A.; Starmer, J.; Wilson, E.M.; Su, M.A. Sex bias in CNS autoimmune disease mediated by androgen control of autoimmune regulator. Nat. Commun. 2016, 7, 11350.
- Dragin, N.; Bismuth, J.; Cizeron-Clairac, G.; Biferi, M.G.; Berthault, C.; Serraf, A.; Nottin, R.; Klatzmann, D.; Cumano, A.; Barkats, M.; et al. Estrogen-mediated downregulation of AIRE influences sexual dimorphism in autoimmune diseases. J. Clin. Invest. 2016, 126, 1525–1537.
- Shi, L.; Hu, L.H.; Li, Y.R. Autoimmune regulator regulates autophagy in THP-1 human monocytes. Front. Med. China 2010, 4, 336–341.
- Rodrigues, P.M.; Sousa, L.G.; Perrod, C.; Maceiras, A.R.; Ferreirinha, P.; Pombinho, R.; Romera-Cárdenas, G.; Gomez-Lazaro, M.; Senkara, M.; Pistolic, J.; et al. LAMP2 regulates autophagy in the thymic epithelium and thymic stroma-dependent CD4 T cell development. Autophagy 2023, 19, 426–439.
- Alessandrini, F.; Pezzè, L.; Ciribilli, Y. LAMPs: Shedding light on cancer biology. Semin. Oncol. 2017, 44, 239–253.
- Liu, S.-P.; Li, X.-M.; Liu, D.-M.; Xie, S.-H.; Zhang, S.-B.; Li, Y.; Xie, Z.-F. LAMP2 as a Biomarker Related to Prognosis and Immune Infiltration in Esophageal Cancer and Other Cancers: A Comprehensive Pan-Cancer Analysis. Front. Oncol. 2022, 12, 884448.
- Kravtsov, D.S.; Erbe, A.K.; Sondel, P.M.; Rakhmilevich, A.L. Roles of CD4+ T cells as mediators of antitumor immunity. Front. Immunol. 2022, 13, 972021.
- Sakowska, J.; Arcimowicz, Ł.; Jankowiak, M.; Papak, I.; Markiewicz, A.; Dziubek, K.; Kurkowiak, M.; Kote, S.; Kaźmierczak-Siedlecka, K.; Połom, K.; et al. Autoimmunity and Cancer-Two Sides of the Same Coin. Front. Immunol. 2022, 13, 793234.
- Greisen, S.R.; Aspari, M.; Deleuran, B. Co-inhibitory molecules – their role in health and autoimmunity; highlighted by immune related adverse events. Front. Immunol. 2022, 13, 883733.
- Cubas, R.; Khan, Z.; Gong, Q.; Moskalenko, M.; Xiong, H.; Ou, Q.; Pai, C.; Rodriguez, R.; Cheung, J.; Chan, A.C. Autoimmunity linked protein phosphatase PTPN22 as a target for cancer immunotherapy. J. Immunother. Cancer 2020, 8, e001439.
- Arechiga, A.F.; Habib, T.; He, Y.; Zhang, X.; Zhang, Z.Y.; Funk, A.; Buckner, J.H. Cutting edge: The PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J. Immunol. 2009, 182, 3343–3347.
- Begovich, A.B.; Carlton, V.E.; Honigberg, L.A.; Schrodi, S.J.; Chokkalingam, A.P.; Alexander, H.C.; Ardlie, K.G.; Huang, Q.; Smith, A.M.; Spoerke, J.M.; et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 2004, 75, 330–337.
- Velaga, M.R.; Wilson, V.; Jennings, C.E.; Owen, C.J.; Herington, S.; Donaldson, P.T.; Ball, S.G.; James, R.A.; Quinton, R.; Perros, P.; et al. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves’ disease. J. Clin. Endocrinol. Metab. 2004, 89, 5862–5865.
- Smyth, D.; Cooper, J.D.; Collins, J.E.; Heward, J.M.; Franklyn, J.A.; Howson, J.M.; Vella, A.; Nutland, S.; Rance, H.E.; Maier, L.; et al. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes 2004, 53, 3020–3023.
- Bottini, N.; Musumeci, L.; Alonso, A.; Rahmouni, S.; Nika, K.; Rostamkhani, M.; MacMurray, J.; Meloni, G.F.; Lucarelli, P.; Pellecchia, M.; et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 2004, 36, 337–338.
- Ho, W.J.; Croessmann, S.; Lin, J.; Phyo, Z.H.; Charmsaz, S.; Danilova, L.; Mohan, A.A.; Gross, N.E.; Chen, F.; Dong, J.; et al. Systemic inhibition of PTPN22 augments anticancer immunity. J. Clin. Invest. 2021, 131, e146950.
- Martyna, B.; Małgorzata, M.W.; Nikola, Z.; Beniamin, G.; Urszula, M.; Grażyna, J. Expression Profile of Genes Associated with the Proteins Degradation Pathways in Colorectal adenocarcinoma. Curr. Pharm. Biotechnol. 2019, 20, 551–561.
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol. 2015, 294, 63–69.
- Maglione, A.; Rolla, S.; Mercanti, S.F.; Cutrupi, S.; Clerico, M. The Adaptive Immune System in Multiple Sclerosis: An Estrogen-Mediated Point of View. Cells 2019, 8, 1280.
- Tai, P.; Wang, J.; Jin, H.; Song, X.; Yan, J.; Kang, Y.; Zhao, L.; An, X.; Du, X.; Chen, X.; et al. Induction of regulatory T cells by physiological level estrogen. J. Cell Physiol. 2008, 214, 456–464.
- Hill, L.; Jeganathan, V.; Chinnasamy, P.; Grimaldi, C.; Diamond, B. Differential roles of estrogen receptors α and β in control of B-cell maturation and selection. Mol. Med. 2011, 17, 211–220.
- Williams, M.M.; Spoelstra, N.S.; Arnesen, S.; O’Neill, K.I.; Christenson, J.L.; Reese, J.; Torkko, K.C.; Goodspeed, A.; Rosas, E.; Hanamura, T.; et al. Steroid Hormone Receptor and Infiltrating Immune Cell Status Reveals Therapeutic Vulnerabilities of ESR1-Mutant Breast Cancer. Cancer Res. 2021, 81, 732–746.
- Dou, C.; Ding, N.; Zhao, C.; Hou, T.; Kang, F.; Cao, Z.; Liu, C.; Bai, Y.; Dai, Q.; Ma, Q.; et al. Estrogen Deficiency-Mediated M2 Macrophage Osteoclastogenesis Contributes to M1/M2 Ratio Alteration in Ovariectomized Osteoporotic Mice. J. Bone. Miner. Res. 2018, 33, 899–908.
- Campbell, L.; Emmerson, E.; Williams, H.; Saville, C.R.; Krust, A.; Chambon, P.; Mace, K.A.; Hardman, M.J. Estrogen receptor-alpha promotes alternative macrophage activation during cutaneous repair. J. Investig. Dermatol. 2014, 134, 2447–2457.
- Ranhotra, H.S. Estrogen-related receptor alpha in select host functions and cancer: New frontiers. Mol. Cell Biochem. 2022, 477, 1349–1359.
- Xiang, J.; Liu, X.; Ren, J.; Chen, K.; Wang, H.L.; Miao, Y.Y.; Qi, M.M. How does estrogen work on autophagy? Autophagy 2019, 15, 197–211.
- De Sousa Linhares, A.; Leitner, J.; Grabmeier-Pfistershammer, K.; Steinberger, P. Not All Immune Checkpoints Are Created Equal. Front. Immunol. 2018, 9, 1909.
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547.
- Wong, C.K.; Lam, T.H.; Liao, S.Y.; Lau, Y.M.; Tse, H.F.; So, B.Y.F. Immunopathogenesis of Immune Checkpoint Inhibitor Induced Myocarditis: Insights from Experimental Models and Treatment Implications. Biomedicines 2023, 11, 107.
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151.
- Oya, Y.; Watanabe, N.; Kobayashi, Y.; Owada, T.; Oki, M.; Ikeda, K.; Suto, A.; Kagami, S.; Hirose, K.; Kishimoto, T.; et al. Lack of B and T lymphocyte attenuator exacerbates autoimmune disorders and induces Fas-independent liver injury in MRL-lpr/lpr mice. Int. Immunol. 2011, 23, 335–344.
- Wang, L.; Le Mercier, I.; Putra, J.; Chen, W.; Liu, J.; Schenk, A.D.; Nowak, E.C.; Suriawinata, A.A.; Li, J.; Noelle, R.J. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc. Natl. Acad. Sci. USA 2014, 111, 14846–14851.
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342.
- Dörner, T.; Szelinski, F.; Lino, A.C.; Lipsky, P.E. Therapeutic implications of the anergic/postactivated status of B cells in systemic lupus erythematosus. RMD Open 2020, 6, e001258.
- Yu, L.; Shao, M.; Zhou, T.; Xie, H.; Wang, F.; Kong, J.; Xu, S.; Shuai, Z.; Pan, F. Association of CTLA-4 (+49 A/G) polymorphism with susceptibility to autoimmune diseases: A meta-analysis with trial sequential analysis. Int. Immunopharmacol. 2021, 96, 107617.
- Chen, S.; Li, Y.; Deng, C.; Li, J.; Wen, X.; Wu, Z.; Hu, C.; Zhang, S.; Li, P.; Zhang, X.; et al. The associations between PD-1, CTLA-4 gene polymorphisms and susceptibility to ankylosing spondylitis: A meta-analysis and systemic review. Rheumatol Int. 2016, 36, 33–44.
- Liu, R.; Wang, X.; Chen, X.; Wang, S.; Zhang, H. TIM-3 rs1036199 polymorphism increases susceptibility to autoimmune diseases: Evidence based on 4200 subjects. Biosci. Rep. 2018, 38, BSR20181235.
- Pawlak-Adamska, E.; Nowak, O.; Karabon, L.; Pokryszko-Dragan, A.; Partyka, A.; Tomkiewicz, A.; Ptaszkowski, J.; Frydecka, I.; Podemski, R.; Dybko, J. Bilinska MPD-1 gene polymorphic variation is linked with first symptom of disease severity of relapsing-remitting form of MS. J. Neuroimmunol. 2017, 305, 115–127.
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26.
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86, a molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603.
- Jain, N.; Nguyen, H.; Chambers, C.; Kang, J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl. Acad. Sci. USA 2010, 107, 1524–1528.
- Verma, N.; Burns, S.O.; Walker, L.S.K.; Sansom, D.M. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin. Exp. Immunol. 2017, 190, 1–7.
- Zhang, B.; Dang, J.; Ba, D.; Wang, C.; Han, J.; Zheng, F. Potential function of CTLA-4 in the tumourigenic capacity of melanoma stem cells. Oncol. Lett. 2018, 16, 6163–6170.
- Roncella, S.; Laurent, S.; Fontana, V.; Ferro, P.; Franceschini, M.C.; Salvi, S.; Varesano, S.; Boccardo, S.; Vigani, A.; Morabito, A.; et al. CTLA-4 in mesothelioma patients: Tissue expression, body fluid levels and possible relevance as a prognostic factor. Cancer Immunol. Immunother. 2016, 65, 909–917.
- Zhao, Y.; Yang, W.; Huang, Y.; Cui, R.; Li, X.; Li, B. Evolving Roles for Targeting CTLA-4 in Cancer Immunotherapy. Cell Physiol. Biochem. 2018, 47, 721–734.
- Urbano, A.C.; Nascimento, C.; Soares, M.; Correia, J.; Ferreira, F. Clinical Relevance of the serum CTLA-4 in Cats with Mammary Carcinoma. Sci. Rep. 2020, 10, 3822.
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145.
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete Foxp3 Þ Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019, 25, 1233–1238.
- Shukla, S.A.; Bachireddy, P.; Schilling, B.; Galonska, C.; Zhan, Q.; Bango, C.; Langer, R.; Lee, P.C.; Gusenleitner, D.; Keskin, D.B.; et al. Cancer-Germline Antigen Expression Discriminates Clinical Outcome to CTLA-4 Blockade. Cell 2018, 173, 624–633.
- Jiang, G.M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.T.; Meng, X.J.; Li, L.L.; Liu, Y.; Li, W.F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 17.
- Pauken, K.E.; Jenkins, M.K.; Azuma, M.; Fife, B.T. PD-1, but not PD-L1, expressed by islet-reactive CD4+ T cells suppresses infiltration of the pancreas during type 1 diabetes. Diabetes 2013, 62, 2859–2869.
- Ke, Y.; Sun, D.; Jiang, G.; Kaplan, H.J.; Shao, H. PD-L1(hi) retinal pigment epithelium (RPE) cells elicited by inflammatory cytokines induce regulatory activity in uveitogenic T cells. J. Leukoc. Biol. 2010, 88, 1241–1249.
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895.
- Colli, M.L.; Hill, J.L.E.; Marroquí, L.; Chaffey, J.; Dos Santos, R.S.; Leete, P.; Coomans de Brachène, A.; Paula, F.M.M.; Op de Beeck, A.; Castela, A.; et al. PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine 2018, 36, 367–375.
- Osum, K.C.; Burrack, A.L.; Martinov, T.; Sahli, N.L.; Mitchell, J.S.; Tucker, C.G.; Pauken, K.E.; Papas, K.; Appakalai, B.; Spanier, J.A.; et al. Interferon-gamma drives programmed death-ligand 1 expression on islet β cells to limit T cell function during autoimmune diabetes. Sci. Rep. 2018, 8, 8295.
- Abdeladhim, M.; Karnell, J.L.; Rieder, S.A. In or out of control: Modulating regulatory T cell homeostasis and function with immune checkpoint pathways. Front. Immunol. 2022, 13, 1033705.
- Zhao, Z.; Wang, X.; Bao, X.Q.; Ning, J.; Shang, M.; Zhang, D. Autoimmune polyendocrine syndrome induced by immune checkpoint inhibitors: A systematic review. Cancer Immunol. Immunother. 2021, 70, 1527–1540.
- Schneider, S.; Potthast, S.; Komminoth, P.; Schwegler, G.; Böhm, S. PD-1 Checkpoint Inhibitor Associated Autoimmune Encephalitis. Case Rep. Oncol. 2017, 10, 473–478.
- Hakroush, S.; Tampe, B. Association between Loss of Immune Checkpoint Programmed Cell Death Protein 1 and Active ANCA-Associated Renal Vasculitis. Int. J. Mol. Sci. 2023, 24, 2975.
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553.
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692.
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer. 2019, 18, 10.
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386.
- Jancewicz, I.; Szarkowska, J.; Konopinski, R.; Stachowiak, M.; Swiatek, M.; Blachnio, K.; Kubala, S.; Oksinska, P.; Cwiek, P.; Rusetska, N.; et al. PD-L1 Overexpression, SWI/SNF Complex Deregulation, and Profound Transcriptomic Changes Characterize Cancer-Dependent Exhaustion of Persistently Activated CD4+ T Cells. Cancers 2021, 13, 4148.
- He, Q.F.; Xu, Y.; Li, J.; Huang, Z.M.; Li, X.H.; Wang, X. CD8+ T-cell exhaustion in cancer: Mechanisms and new area for cancer immunotherapy. Brief. Funct. Genom. 2019, 18, 99–106.
- Robainas, M.; Otano, R.; Bueno, S.; Ait-Oudhia, S. Understanding the role of PD-L1/PD1 pathway blockade and autophagy in cancer therapy. OncoTargets Ther. 2017, 10, 1803–1807.
- Maher, C.M.; Thomas, J.D.; Haas, D.A.; Longen, C.G.; Oyer, H.M.; Tong, J.Y.; Kim, F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2018, 16, 243–255.
- Ashrafizadeh, M.; Zarrabi, A.; Hushmandi, K.; Zarrin, V.; Moghadam, E.R.; Zabolian, A.; Tavakol, S.; Samarghandian, S.; Najafi, M. PD-1/PD-L1 axis regulation in cancer therapy: The role of long non-coding RNAs and microRNAs. Life Sci. 2020, 256, 117899.
- Clark, C.A.; Gupta, H.B.; Curiel, T.J. Tumor cell-intrinsic CD274/PD-L1, A novel metabolic balancing act with clinical potential. Autophagy 2017, 13, 987–988.
- Cai, J.; Qi, Q.; Qian, X.; Han, J.; Zhu, X.; Zhang, Q.; Xia, R. The role of PD-1/PD-L1 axis and macrophage in the progression and treatment of cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 1377–1385.
- Yamada, A.; Arakaki, R.; Saito, M.; Kudo, Y.; Ishimaru, N. Dual Role of Fas/FasL-Mediated Signal in Peripheral Immune Tolerance. Front. Immunol. 2017, 8, 403.
- Niederkorn, J.Y. See no evil, hear no evil, do no evil: The lessons of immune privilege. Nat. Immunol. 2006, 7, 354–359.
- Li, H.; Tsokos, G.C. Double-negative T cells in autoimmune diseases. Curr. Opin. Rheumatol. 2021, 33, 163–172.
- Rensing-Ehl, A.; Völkl, S.; Speckmann, C.; Lorenz, M.R.; Ritter, J.; Janda, A.; Abinun, M.; Pircher, H.; Bengsch, B.; Thimme, R.; et al. Abnormally differentiated CD4+ or CD8+ T cells with phenotypic and genetic features of double negative T cells in human Fas deficiency. Blood 2014, 124, 851–860.
- Saxena, A.; Yagita, H.; Donner, T.W.; Hamad, A.R.A. Expansion of FasL-Expressing CD5+ B Cells in Type 1 Diabetes Patients. Front. Immunol. 2017, 8, 402.
- Li, L.; Liu, S.; Yu, J. Autoimmune thyroid disease and type 1 diabetes mellitus: Same pathogenesis; new perspective? Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820958329.
- de Oliveira, G.L.; Ferreira, A.F.; Gasparotto, E.P.; Kashima, S.; Covas, D.T.; Guerreiro, C.T.; Brum, D.G.; Barreira, A.A.; Voltarelli, J.C.; Simões, B.P.; et al. Defective expression of apoptosis-related molecules in multiple sclerosis patients is normalized early after autologous haematopoietic stem cell transplantation. Clin. Exp. Immunol. 2017, 187, 383–398.
- Hohlbaum, A.M.; Moe, S.; Marshak-Rothstein, A. Opposing effects of transmembrane and soluble Fas ligand expression on inflammation and tumor cell survival. J. Exp. Med. 2000, 191, 1209–1220.
- Vincent, F.B.; Kandane-Rathnayake, R.; Koelmeyer, R.; Harris, J.; Hoi, A.Y.; Mackay, F.; Morand, E.F. Associations of serum soluble Fas and Fas ligand (FasL) with outcomes in systemic lupus erythematosus. Lupus Sci. Med. 2020, 7, e000375.
- Vincent, F.B.; Bubicich, M.; Downie-Doyle, S.; Mackay, F.; Morand, E.F.; Rischmueller, M. Serum soluble Fas and Fas ligand (FasL) in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S118), 254–256.
- Horton, B.L.; Williams, J.B.; Cabanov, A.; Spranger, S.; Gajewski, T.F. Intratumoral CD8+ T-cell Apoptosis Is a Major Component of T-cell Dysfunction and Impedes Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 14–24.
- Zhu, J.; Powis de Tenbossche, C.G.; Cané, S.; Colau, D.; van Baren, N.; Lurquin, C.; Schmitt-Verhulst, A.M.; Liljeström, P.; Uyttenhove, C.; Van den Eynde, B.J. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat. Commun. 2017, 8, 1404.
- Wada, A.; Tada, Y.; Kawamura, K.; Takiguchi, Y.; Tatsumi, K.; Kuriyama, T.; Takenouchi, T.; O-Wang, J.; Tagawa, M. The effects of FasL on inflammation and tumor survival are dependent on its expression levels. Cancer Gene Ther. 2007, 14, 262–267.
- Khodapasand, E.; Jafarzadeh, N.; Farrokhi, F.; Kamalidehghan, B.; Houshmand, M. Is Bax/Bcl-2 ratio considered as a prognostic marker with age and tumor location in colorectal cancer? Iran Biomed. J. 2015, 19, 69–75.
- Kulsoom, B.; Shamsi, T.S.; Afsar, N.A.; Memon, Z.; Ahmed, N.; Hasnain, S.N. Bax, Bcl-2, and Bax/Bcl-2 as prognostic markers in acute myeloid leukemia: Are we ready for Bcl-2-directed therapy? Cancer Manag. Res. 2018, 10, 403–416.
- Raisova, M.; Hossini, A.M.; Eberle, J.; Riebeling, C.; Wieder, T.; Sturm, I.; Daniel, P.T.; Orfanos, C.E.; Geilen, C.C. The Bax/Bcl-2 ratio determines the susceptibility of human melanoma cells to CD95/Fas-mediated apoptosis. J. Investig. Dermatol. 2001, 117, 333–340.
- Ploumaki, I.; Triantafyllou, E.; Koumprentziotis, I.A.; Karampinos, K.; Drougkas, K.; Karavolias, I.; Trontzas, I.; Kotteas, E.A. Bcl-2 pathway inhibition in solid tumors: A review of clinical trials. Clin. Transl. Oncol. 2023, 1–25.
- Mérino, D.; Bouillet, P. The Bcl-2 family in autoimmune and degenerative disorders. Apoptosis 2009, 14, 570–583.
- Strasser, A.; Harris, A.W.; Cory, S. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell 1991, 67, 889–899.
- Strasser, A.; Whittingham, S.; Vaux, D.L.; Bath, M.L.; Adams, J.M.; Cory, S.; Harris, A.W. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc. Natl. Acad. Sci. USA 1991, 88, 8661–8665.
- Li, M.; Gao, P.; Zhang, J. Crosstalk between Autophagy and Apoptosis: Potential and Emerging Therapeutic Targets for Cardiac Diseases. Int. J. Mol. Sci. 2016, 17, 332.
- Chen, Y.; Liu, X.R.; Yin, Y.Q.; Lee, C.J.; Wang, F.T.; Liu, H.Q.; Wu, X.T.; Liu, J. Unravelling the multifaceted roles of Atg proteins to improve cancer therapy. Cell Prolif. 2014, 47, 105–112.
- Hounsell, C.; Fan, Y. The Duality of Caspases in Cancer, as Told through the Fly. Int. J. Mol. Sci. 2021, 22, 8927.
- You, R.; He, X.; Zeng, Z.; Zhan, Y.; Xiao, Y.; Xiao, R. Pyroptosis and Its Role in Autoimmune Disease: A Potential Therapeutic Target. Front. Immunol. 2022, 13, 841732.
- Pezone, A.; Olivieri, F.; Napoli, M.V.; Procopio, A.; Avvedimento, E.V.; Gabrielli, A. Inflammation and DNA damage: Cause, effect or both. Nat. Rev. Rheumatol. 2023, 19, 200–211.
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974.
- Kataoka, T. The caspase-8 modulator c-FLIP. Crit. Rev. Immunol. 2005, 25, 31–58.
- Seki, M.; Oomizu, S.; Sakata, K.M.; Sakata, A.; Arikawa, T.; Watanabe, K.; Ito, K.; Takeshita, K.; Niki, T.; Saita, N.; et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin. Immunol. 2008, 127, 78–88.
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252.
- Liu, X.; Alexiou, M.; Martin-Orozco, N.; Chung, Y.; Nurieva, R.I.; Ma, L.; Tian, Q.; Kollias, G.; Lu, S.; Graf, D.; et al. Cutting edge: A critical role of B and T lymphocyte attenuator in peripheral T cell tolerance induction. J. Immunol. 2009, 182, 4516–4520.
- Wiedemann, A.; Lettau, M.; Weißenberg, S.Y.; Stefanski, A.L.; Schrezenmeier, E.V.; Rincon-Arevalo, H.; Reiter, K.; Alexander, T.; Hiepe, F.; Lino, A.C.; et al. BTLA Expression and Function Are Impaired on SLE B Cells. Front. Immunol. 2021, 12, 667991.
- Oster, C.; Wilde, B.; Specker, C.; Sun, M.; Kribben, A.; Witzke, O.; Dolff, S. BTLA Expression on Th1, Th2 and Th17 Effector T-Cells of Patients with Systemic Lupus Erythematosus Is Associated with Active Disease. Int. J. Mol. Sci. 2019, 20, 4505.
- Piancone, F.; Saresella, M.; Marventano, I.; La Rosa, F.; Zoppis, M.; Agostini, S.; Longhi, R.; Caputo, D.; Mendozzi, L.; Rovaris, M.; et al. B Lymphocytes in Multiple Sclerosis: Bregs and BTLA/CD272 Expressing-CD19+ Lymphocytes Modulate Disease Severity. Sci. Rep. 2016, 6, 29699.
- Liu, J.; Ming, S.; Song, W.; Meng, X.; Xiao, Q.; Wu, M.; Wu, Y.; Xie, H.; Zhou, J.; Zhong, H.; et al. B and T lymphocyte attenuator regulates autophagy in mycobacterial infection via the AKT/mTOR signal pathway. Int. Immunopharmacol. 2021, 91, 107215.
- Cheng, T.Y.; Liu, Y.J.; Yan, H.; Xi, Y.B.; Duan, L.Q.; Wang, Y.; Zhang, T.T.; Gu, Y.M.; Wang, X.D.; Wu, C.X.; et al. Tumor Cell-Intrinsic BTLA Receptor Inhibits the Proliferation of Tumor Cells via ERK1/2. Cells 2022, 11, 4021.
- Chen, M.M.; Xiao, X.; Lao, X.M.; Wei, Y.; Liu, R.X.; Zeng, Q.H.; Wang, J.C.; Ouyang, F.Z.; Chen, D.P.; Chan, K.W.; et al. Polarization of Tissue-Resident TFH-Like Cells in Human Hepatoma Bridges Innate Monocyte Inflammation and M2b Macrophage Polarization. Cancer Discov. 2016, 6, 1182–1195.
- Wu, H.; Tang, S.; Zhou, M.; Xue, J.; Yu, Z.; Zhu, J. Tim-3 suppresses autoimmune hepatitis via the p38/MKP-1 pathway in Th17 cells. FEBS Open Bio. 2021, 11, 1406–1416.
- Schatton, T.; Itoh, Y.; Martins, C.; Rasbach, E.; Singh, P.; Silva, M.; Mucciarone, K.; Heppt, M.V.; Geddes-Sweeney, J.; Stewart, K.; et al. Inhibition of Melanoma Cell-Intrinsic Tim-3 Stimulates MAPK-Dependent Tumorigenesis. Cancer Res. 2022, 82, 3774–3784.
- Lee, C.S.; Yuan, T.L.; Chakka, S.; Fellmann, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517.
- Fuhrman, C.A.; Yeh, W.I.; Seay, H.R.; Saikumar Lakshmi, P.; Chopra, G.; Zhang, L.; Perry, D.J.; McClymont, S.A.; Yadav, M.; Lopez, M.C.; et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J. Immunol. 2015, 195, 145–155.
- Wang, N.; Liang, S.; Jin, J.; Fang, L.; Ma, Q.; Wang, X.; Song, Y. Chen LCD226 attenuates Treg suppressive capacity via, C.T.L.A.-4.; TIGIT during, E.A.E. Immunol. Res. 2019, 67, 486–496.
- Zhang, J.; Zhou, L.; Xiang, J.D.; Jin, C.S.; Li, M.Q.; He, Y.Y. Artesunate-induced ATG5-related autophagy enhances the cytotoxicity of NK92 cells on endometrial cancer cells via interactions between CD155 and CD226/TIGIT. Int. Immunopharmacol. 2021, 97, 107705.
- Reches, A.; Ophir, Y.; Stein, N.; Kol, I.; Isaacson, B.; Charpak-Amikam, Y.; Elnekave, A.; Tsukerman, P.; Kucan Brlic, P.; Lenac, T.; et al. Nectin4 is a Novel TIGIT Ligand Which Combines Checkpoint Inhibition and Tumor Specificity. J. Immunother. Cancer 2020, 8, e000266.
- Yang, B.; Xue, Q.; Guo, J.; Wang, X.; Zhang, Y.; Guo, K.; Li, W.; Chen, S.; Xue, T.; Qi, X.; et al. Autophagy induction by the pathogen receptor NECTIN4 and sustained autophagy contribute to peste des petits ruminants virus infectivity. Autophagy 2020, 16, 842–861.
- Caramalho, Í.; Nunes-Cabaço, H.; Foxall, R.B.; Sousa, A.E. Regulatory T-Cell Development in the Human Thymus. Front. Immunol. 2015, 6, 395.
- Richards, D.M.; Delacher, M.; Goldfarb, Y.; Kägebein, D.; Hofer, A.C.; Abramson, J.; Feuerer, M. Treg Cell Differentiation: From Thymus to Peripheral Tissue. Prog. Mol. Biol. Transl. Sci. 2015, 136, 175–205.
- Ryba-Stanisławowska, M.; Sakowska, J.; Zieliński, M.; Ławrynowicz, U.; Trzonkowski, P. Regulatory T cells: The future of autoimmune disease treatment. Expert Rev. Clin. Immunol. 2019, 15, 777–789.
- Maceiras, A.R.; Almeida, S.C.P.; Mariotti-Ferrandiz, E.; Chaara, W.; Jebbawi, F.; Six, A.; Hori, S.; Klatzmann, D.; Faro, J.; Graca, L. T follicular helper and T follicular regulatory cells have different TCR specificity. Nat. Commun. 2017, 8, 15067.
- Vaeth, M.; Müller, G.; Stauss, D.; Dietz, L.; Klein-Hessling, S.; Serfling, E.; Lipp, M.; Berberich, I.; Berberich-Siebelt, F. Follicular regulatory T cells control humoral autoimmunity via NFAT2-regulated CXCR5 expression. J. Exp. Med. 2014, 211, 545–561.
- Fisson, S.; Darrasse-Jèze, G.; Litvinova, E.; Septier, F.; Klatzmann, D.; Liblau, R.; Salomon, B.L. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J. Exp. Med. 2003, 198, 737–746.
- Lewkowicz, N.; Klink, M.; Mycko, M.P.; Lewkowicz, P. Neutrophil-CD4+CD25+ T regulatory cell interactions: A possible new mechanism of infectious tolerance. Immunobiology 2013, 218, 455–464.
- Piekarska, K.; Urban-Wójciuk, Z.; Kurkowiak, M.; Pelikant-Małecka, I.; Schumacher, A.; Sakowska, J.; Spodnik, J.H.; Arcimowicz, Ł.; Zielińska, H.; Tymoniuk, B.; et al. Mesenchymal stem cells transfer mitochondria to allogeneic Tregs in an HLA-dependent manner improving their immunosuppressive activity. Nat. Commun. 2022, 13, 856.
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208.
- Sullivan, J.A.; Tomita, Y.; Jankowska-Gan, E.; Lema, D.A.; Arvedson, M.P.; Nair, A.; Bracamonte-Baran, W.; Zhou, Y.; Meyer, K.K.; Zhong, W.; et al. Treg-Cell-Derived IL-35-Coated Extracellular Vesicles Promote Infectious Tolerance. Cell Rep. 2020, 30, 1039–1051.e5.
- Barzaghi, F.; Passerini, L. IPEX Syndrome: Improved Knowledge of Immune Pathogenesis Empowers Diagnosis. Front. Pediatr. 2021, 9, 612760.
- Wildin, R.S.; Ramsdell, F.; Peake, J.; Faravelli, F.; Casanova, J.L.; Buist, N.; Levy-Lahad, E.; Mazzella, M.; Goulet, O.; Perroni, L.; et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001, 27, 18–20.
- de Kleer, I.M.; Wedderburn, L.R.; Taams, L.S.; Patel, A.; Varsani, H.; Klein, M.; de Jager, W.; Pugayung, G.; Giannoni, F.; Rijkers, G.; et al. CD4+CD25bright regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J. Immunol. 2004, 172, 6435–6443.
- van Amelsfort, J.M.; Jacobs, K.M.; Bijlsma, J.W.; Lafeber, F.P.; Taams, L.S. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: Differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. 2004, 50, 2775–2785.
- Slobodin, G.; Ahmad, M.S.; Rosner, I.; Peri, R.; Rozenbaum, M.; Kessel, A.; Toubi, E.; Odeh, M. Regulatory T cells (CD4(+)CD25(bright)FoxP3(+)) expansion in systemic sclerosis correlates with disease activity and severity. Cell Immunol. 2010, 261, 77–80.
- La Cava, A. Tregs in SLE: An Update. Curr. Rheumatol. Rep. 2018, 20, 6.
- Vitales-Noyola, M.; Serrano-Somavilla, A.; Martínez-Hernández, R.; Sampedro-Nuñez, M.; Ramos-Levi, A.M.; González-Amaro, R.; Marazuela, M. Patients with Autoimmune Thyroiditis Show Diminished Levels and Defective Suppressive Function of Tr1 Regulatory Lymphocytes. J. Clin. Endocrinol. Metab. 2018, 103, 3359–3367.
- Marazuela, M.; García-López, M.A.; Figueroa-Vega, N.; De La Fuente, H.; Alvarado, B.; Monsiváis-Urenda, A.; Sánchez-Madrid, F.; González-Amaro, R. Regulatory T Cells in Human Autoimmune Thyroid Disease. J. Clin. Endocrinol. Metab. 2006, 91, 3639–3646.
- Kumar, M.; Putzki, N.; Limmroth, V.; Remus, R.; Lindemann, M.; Knop, D.; Mueller, N.; Hardt, C.; Kreuzfelder, E.; Grosse-Wilde, H. CD4+CD25+FoxP3+ T lymphocytes fail to suppress myelin basic protein-induced proliferation in patients with multiple sclerosis. J. Neuroimmunol. 2006, 180, 178–184.
- Venken, K.; Hellings, N.; Thewissen, M.; Somers, V.; Hensen, K.; Rummens, J.L.; Medaer, R.; Hupperts, R.; Stinissen, P. Compromised CD4+ CD25(high) regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology 2008, 123, 79–89.
- Holohan, D.R.; Van Gool, F.; Bluestone, J.A. Thymically-derived Foxp3+ regulatory T cells are the primary regulators of type 1 diabetes in the non-obese diabetic mouse model. PLoS ONE 2019, 14, e0217728.
- Bending, D.; Giannakopoulou, E.; Lom, H.; Wedderburn, L.R. Synovial Regulatory T Cells Occupy a Discrete TCR Niche in Human Arthritis and Require Local Signals To Stabilize FOXP3 Protein Expression. J. Immunol. 2015, 195, 5616–5624.
- Yang, S.; Zhang, X.; Chen, J.; Dang, J.; Liang, R.; Zeng, D.; Zhang, H.; Xue, Y.; Liu, Y.; Wu, W.; et al. Induced, But Not Natural, Regulatory T Cells Retain Phenotype and Function Following Exposure to Inflamed Synovial Fibroblasts. Sci. Adv. 2020, 6, eabb0606.
- Pesenacker, A.M.; Bending, D.; Ursu, S.; Wu, Q.; Nistala, K.; Wedderburn, L. CD161 defines the subset of FoxP3+ T cells capable of producing proinflammatory cytokines. Blood 2013, 121, 2647–2658.
- Qiu, R.; Zhou, L.; Ma, Y.; Zhou, L.; Liang, T.; Shi, L.; Long, J.; Yuan, D. Regulatory T Cell Plasticity and Stability and Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2020, 58, 52–70.
- Yang, X.O.; Nurieva, R.; Martinez, G.J.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008, 29, 44–56.
- Zhang, J.; Chen, L.; Xiong, F.; Zhang, S.; Huang, K.; Zhang, Z.; Wang, C.Y. Autophagy in regulatory T cells: A double-edged sword in disease settings. Mol. Immunol. 2019, 109, 43–50.
- Alissafi, T.; Banos, A.; Boon, L.; Sparwasser, T.; Ghigo, A.; Wing, K.; Vassilopoulos, D.; Boumpas, D.; Chavakis, T.; Cadwell, K.; et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J. Clin. Invest. 2017, 127, 2789–2804.
- de Oliveira, C.E.; Gasparoto, T.H.; Pinheiro, C.R.; Amôr, N.G.; Nogueira, M.R.S.; Kaneno, R.; Garlet, G.P.; Lara, V.S.; Silva, J.S.; Cavassani, K.A.; et al. CCR5-Dependent Homing of T Regulatory Cells to the Tumor Microenvironment Contributes to Skin Squamous Cell Carcinoma Development. Mol. Cancer Ther. 2017, 16, 2871–2880.
- Mizukami, Y.; Kono, K.; Kawaguchi, Y.; Akaike, H.; Kamimura, K.; Sugai, H.; Fujii, H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int. J. Cancer. 2008, 122, 2286–2293.
- Lu, L.; Ma, J.; Li, Z.; Lan, Q.; Chen, M.; Liu, Y.; Xia, Z.; Wang, J.; Han, Y.; Shi, W.; et al. All-trans retinoic acid promotes TGF-β-induced Tregs via histone modification but not DNA demethylation on Foxp3 gene locus. PLoS ONE 2011, 6, e24590.
- Shitara, K.; Nishikawa, H. Regulatory T cells: A potential target in cancer immunotherapy. Ann. N. York Acad. Sci. 2018, 1417, 104–115.
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines 2016, 4, 28.
- Que, W.; Guo, W.Z.; Li, X.K. Manipulation of Regulatory Dendritic Cells for Induction Transplantation Tolerance. Front. Immunol. 2020, 11, 582658.
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10, 2689.
- Lindqvist, C.A.; Christiansson, L.H.; Simonsson, B.; Enblad, G.; Olsson-Strömberg, U.; Loskog, A.S. T regulatory cells control T-cell proliferation partly by the release of soluble CD25 in patients with B-cell malignancies. Immunology 2010, 131, 371–376.
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735.
- Turnis, M.E.; Sawant, D.V.; Szymczak-Workman, A.L.; Andrews, L.P.; Delgoffe, G.M.; Yano, H.; Beres, A.J.; Vogel, P.; Workman, C.J.; Vignali, D.A. Interleukin-35 Limits Anti-Tumor Immunity. Immunity 2016, 44, 316–329.
- Correale, P.; Rotundo, M.S.; Del Vecchio, M.T.; Remondo, C.; Migali, C.; Ginanneschi, C.; Tsang, K.Y.; Licchetta, A.; Mannucci, S.; Loiacono, L.; et al. Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J. Immunother. 2010, 33, 435–441.
- Shen, Z.; Zhou, S.; Wang, Y.; Li, R.L.; Zhong, C.; Liang, C.; Sun, Y. Higher intratumoral infiltrated Foxp3+ Treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 1585–1595.
- Bonertz, A.; Weitz, J.; Pietsch, D.H.; Rahbari, N.N.; Schlude, C.; Ge, Y.; Juenger, S.; Vlodavsky, I.; Khazaie, K.; Jaeger, D.; et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J. Clin. Investig. 2009, 119, 3311–3321.
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285.
- Le Texier, L.; Lineburg, K.E.; Cao, B.; McDonald-Hyman, C.; Leveque-El Mouttie, L.; Nicholls, J.; Melino, M.; Nalkurthi, B.C.; Alexander, K.A.; Teal, B.; et al. Autophagy-dependent regulatory T cells are critical for the control of graft-versus-host disease. JCI Insight 2016, 1, e86850.
- Duong, B.H.; Ota, T.; Aoki-Ota, M.; Cooper, A.B.; Ait-Azzouzene, D.; Vela, J.L.; Gavin, A.L.; Nemazee, D. Negative selection by IgM superantigen defines a B cell central tolerance compartment and reveals mutations allowing escape. J. Immunol. 2011, 187, 5596–5605.
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294.
- Zhao, Y.; Shen, M.; Feng, Y.; He, R.; Xu, X.; Xie, Y.; Shi, X.; Zhou, M.; Pan, S.; Wang, M.; et al. Regulatory B cells induced by pancreatic cancer cell-derived interleukin-18 promote immune tolerance via the PD-1/PD-L1 pathway. Oncotarget 2017, 9, 14803–14814.
- Ding, Q.; Yeung, M.; Camirand, G.; Zeng, Q.; Akiba, H.; Yagita, H.; Chalasani, G.; Sayegh, M.H.; Najafian, N.; Rothstein, D.M. Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice. J. Clin. Invest. 2011, 121, 3645–3656.
- Baba, Y.; Saito, Y.; Kotetsu, Y. Heterogeneous subsets of B-lineage regulatory cells (Breg cells). Int. Immunol. 2020, 32, 155–162.
- Chen, Y.; Li, C.; Lu, Y.; Zhuang, H.; Gu, W.; Liu, B.; Liu, F.; Sun, J.; Yan, B.; Weng, D.; et al. IL-10-Producing CD1dhiCD5+ Regulatory B Cells May Play a Critical Role in Modulating Immune Homeostasis in Silicosis Patients. Front. Immunol. 2017, 8, 110.
- Noh, J.; Lee, J.H.; Noh, G.; Bang, S.Y.; Kim, H.S.; Choi, W.S.; Cho, S.; Lee, S.S. Characterisation of allergen-specific responses of IL-10-producing regulatory B cells (Br1) in Cow Milk Allergy. Cell Immunol. 2010, 264, 143–149.
- Iwata, Y.; Matsushita, T.; Horikawa, M.; Dilillo, D.J.; Yanaba, K.; Venturi, G.M.; Szabolcs, P.M.; Bernstein, S.H.; Magro, C.M.; Williams, A.D.; et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 2011, 117, 530–541.
- Blair, P.A.; Noreña, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 2010, 32, 129–140.
- Wang, W.; Yuan, X.; Chen, H.; Xie, G.; Ma, Y.; Zheng, Y.; Zhou, Y.; Shen, L. CD19+CD24hiCD38hiBregs involved in downregulate helper T cells and upregulate regulatory T cells in gastric cancer. Oncotarget 2015, 6, 33486–33499.
- Flores-Borja, F.; Bosma, A.; Ng, D.; Reddy, V.; Ehrenstein, M.R.; Isenberg, D.A.; Mauri, C. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci. Transl. Med. 2013, 5, 173ra23.
- van de Veen, W.; Stanic, B.; Yaman, G.; Wawrzyniak, M.; Söllner, S.; Akdis, D.G.; Rückert, B.; Akdis, C.A.; Akdis, M. IgG4 production is confined to human IL-10-producing regulatory B cells that suppress antigen-specific immune responses. J. Allergy Clin. Immunol. 2013, 131, 1204–1212.
- Díaz-Alderete, A.; Crispin, J.C.; Vargas-Rojas, M.I.; Alcocer-Varela, J. IL-10 production in B cells is confined to CD154+ cells in patients with systemic lupus erythematosus. J. Autoimmun. 2004, 23, 379–383.
- Xiao, X.; Lao, X.-M.; Chen, M.-M.; Liu, R.-X.; Wei, Y.; Ouyang, F.-Z.; Chen, D.-P.; Zhao, X.-Y.; Zhao, Q.; Li, X.-F.; et al. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov. 2016, 6, 546–559.
- Matsumoto, M.; Baba, A.; Yokota, T.; Nishikawa, H.; Ohkawa, Y.; Kayama, H.; Kallies, A.; Nutt, S.L.; Sakaguchi, S.; Takeda, K.; et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity 2014, 41, 1040–1051.
- Kessel, A.; Haj, T.; Peri, R.; Snir, A.; Melamed, D.; Sabo, E.; Toubi, E. Human CD19(+)CD25(high) B regulatory cells suppress proliferation of CD4(+) T cells and enhance Foxp3 and CTLA-4 expression in T-regulatory cells. Autoimmun. Rev. 2012, 11, 670–677.
- Matsushita, T. Regulatory and effector B cells: Friends or foes? J. Dermatol. Sci. 2019, 93, 2–7.
- Yoshizaki, A.; Miyagaki, T.; DiLillo, D.J.; Matsushita, T.; Horikawa, M.; Kountikov, E.I.; Spolski, R.; Poe, J.C.; Leonard, W.J.; Tedder, T.F. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature 2012, 491, 264–268.
- Mauri, C.; Gray, D.; Mushtaq, N.; Londei, M. Prevention of arthritis by interleukin 10-producing B cells. J. Exp. Med. 2003, 197, 489–501.
- Watanabe, R.; Ishiura, N.; Nakashima, H.; Kuwano, Y.; Okochi, H.; Tamaki, K.; Sato, S.; Tedder, T.F.; Fujimoto, M. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J. Immunol. 2010, 184, 4801–4809.
- Matsushita, T.; Yanaba, K.; Bouaziz, J.D.; Fujimoto, M.; Tedder, T.F. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Investig. 2008, 118, 3420–3430.
- Lo-Man, R. Regulatory B cells control dendritic cell functions. Immunotherapy 2011, 3 (Suppl. S4), 19–20.
- Yang, M.; Rui, K.; Wang, S.; Lu, L. Regulatory B cells in autoimmune diseases. Cell Mol. Immunol. 2013, 10, 122–132.
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love-hate relationship. Trends Cancer 2016, 2, 747–757.
- Zhou, J.; Min, Z.; Zhang, D.; Wang, W.; Marincola, F.; Wang, X. Enhanced frequency and potential mechanism of B regulatory cells in patients with lung cancer. J. Transl. Med. 2014, 12, 304.
- Murakami, Y.; Saito, H.; Shimizu, S.; Kono, Y.; Shishido, Y.; Miyatani, K.; Matsunaga, T.; Fukumoto, Y.; Ashida, K.; Sakabe, T.; et al. Increased regulatory B cells are involved in immune evasion in patients with gastric cancer. Sci. Rep. 2019, 9, 13083.
- Ishigami, E.; Sakakibara, M.; Sakakibara, J.; Masuda, T.; Fujimoto, H.; Hayama, S.; Nagashima, T.; Sangai, T.; Nakagawa, A.; Nakatani, Y.; et al. Coexistence of regulatory B cells and regulatory T cells in tumor-infiltrating lymphocyte aggregates is a prognostic factor in patients with breast cancer. Breast Cancer 2019, 26, 180–189.
- Lizotte, P.H.; Ivanova, E.V.; Awad, M.M.; Jones, R.E.; Keogh, L.; Liu, H.; Dries, R.; Almonte, C.; Herter-Sprie, G.S.; Santos, A.; et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight. 2016, 1, e89014.
- Zhang, Y.; Morgan, R.; Chen, C.; Cai, Y.; Clark, E.; Khan, W.N.; Shin, S.U.; Cho, H.M.; Al Bayati, A.; Pimentel, A.; et al. Mammary-tumor-educated B cells acquire LAP/TGF-β and PD-L1 expression and suppress anti-tumor immune responses. Int. Immunol. 2016, 28, 423–433.
- Lindner, S.; Dahlke, K.; Sontheimer, K.; Hagn, M.; Kaltenmeier, C.; Barth, T.F.; Beyer, T.; Reister, F.; Fabricius, D.; Lotfi, R.; et al. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer Res. 2013, 73, 2468–2479.
- Bodogai, M.; Moritoh, K.; Lee-Chang, C.; Hollander, C.M.; Sherman-Baust, C.A.; Wersto, R.P.; Araki, Y.; Miyoshi, I.; Yang, L.; Trinchieri, G.; et al. Immunosuppressive and Prometastatic Functions of Myeloid-Derived Suppressive Cells Rely upon Education from Tumor-Associated B Cells. Cancer Res. 2015, 75, 3456–3465.
- Olkhanud, P.B.; Damdinsuren, B.; Bodogai, M.; Gress, R.E.; Sen, R.; Wejksza, K.; Malchinkhuu, E.; Wersto, R.P.; Biragyn, A. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4⁺ T cells to T-regulatory cells. Cancer Res. 2011, 71, 3505–3515.
- Pylayeva-Gupta, Y.; Das, S.; Handler, J.S.; Hajdu, C.H.; Coffre, M.; Koralov, S.B.; Bar-Sagi, D. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 2016, 6, 247–255.
- Zhou, M.; Wen, Z.; Cheng, F.; Ma, J.; Li, W.; Ren, H.; Sheng, Y.; Dong, H.; Lu, L.; Hu, H.M.; et al. Tumor-released autophagosomes induce IL-10-producing B cells with suppressive activity on T lymphocytes via TLR2-MyD88-NF-κB signal pathway. Oncoimmunology 2016, 5, e1180485.
- Corsale, A.M.; Di Simone, M.; Lo Presti, E.; Dieli, F.; Meraviglia, S. γδ T cells and their clinical application in colon cancer. Front. Immunol. 2023, 14, 1098847.
- Nielsen, M.M.; Witherden, D.A.; Havran, W.L. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat. Rev Immunol. 2017, 17, 733–745.
- Mikulak, J.; Oriolo, F.; Bruni, E.; Roberto, A.; Colombo, F.S.; Villa, A.; Bosticardo, M.; Bortolomai, I.; Presti, E.L.; Meraviglia, S.; et al. NKp46-expressing human gut-resident intraepithelial Vδ1 T cell subpopulation exhibits high antitumor activity against colorectal cancer. JCI Insight 2019, 4, e125884.
- Shiromizu, C.M.; Jancic, C.C. γδ T Lymphocytes: An Effector Cell in Autoimmunity and Infection. Front. Immunol. 2018, 9, 2389.
- Isailovic, N.; Daigo, K.; Mantovani, A.; Selmi, C. Interleukin-17 and innate immunity in infections and chronic inflammation. J. Autoimmun. 2015, 60, 1–11.
- Burkett, P.R.; Meyer zu Horste, G.; Kuchroo, V.K. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J. Clin. Investig. 2015, 125, 2211–2219.
- Papotto, P.H.; Reinhardt, A.; Prinz, I.; Silva-Santos, B. Innately versatile: γδ17 T cells in inflammatory and autoimmune diseases. J. Autoimmun. 2018, 87, 26–37.
- Moens, E.; Brouwer, M.; Dimova, T.; Goldman, M.; Willems, F.; Vermijlen, D. IL-23R and TCR signaling drives the generation of neonatal Vgamma9Vdelta2 T cells expressing high levels of cytotoxic mediators and producing IFN-gamma and IL-17. J. Leukoc Biol. 2011, 89, 743–752.
- Doherty, D.G. Immunity, tolerance and autoimmunity in the liver: A comprehensive review. J. Autoimmun. 2016, 66, 60–75.
- Cong, L.H.; Li, T.; Wang, H.; Wu, Y.N.; Wang, S.P.; Zhao, Y.Y.; Zhang, G.Q.; Duan, J. IL-17A-producing T cells exacerbate fine particulate matter-induced lung inflammation and fibrosis by inhibiting PI3K/Akt/mTOR-mediated autophagy. J. Cell Mol. Med. 2020, 24, 8532–8544.
- Wang, Z.; Zhou, H.; Zheng, H.; Zhou, X.; Shen, G.; Teng, X.; Liu, X.; Zhang, J.; Wei, X.; Hu, Z.; et al. Autophagy-based unconventional secretion of HMGB1 by keratinocytes plays a pivotal role in psoriatic skin inflammation. Autophagy 2021, 17, 529–552.
- Silva-Santos, B.; Serre, K.; Norell, H. γδ T cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691.
- Gao, Z.; Bai, Y.; Lin, A.; Jiang, A.; Zhou, C.; Cheng, Q.; Liu, Z.; Chen, X.; Zhang, J.; Luo, P. Gamma delta T-cell-based immune checkpoint therapy: Attractive candidate for antitumor treatment. Mol. Cancer 2023, 22, 31.
- Liu, Y.; Zhang, C. The Role of Human γδ T Cells in Anti-Tumor Immunity and Their Potential for Cancer Immunotherapy. Cells 2020, 9, 1206.
- Tao, L.F.; Yang, B.Q.; Zeng, Z.Y.; Xu, J.P.; Lin, D.H.; Chen, Q.C.; Chen, J.M. Effect of γδ T cells on the Proliferation, Apoptosis and Autophagy of Multiple Myeloma Cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2022, 30, 797–803.
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 2014, 40, 785–800.
- Meraviglia, S.; Lo Presti, E.; Tosolini, M.; La Mendola, C.; Orlando, V.; Todaro, M.; Catalano, V.; Stassi, G.; Cicero, G.; Vieni, S.; et al. Distinctive features of tumor-infiltrating γδ T lymphocytes in human colorectal cancer. Oncoimmunology 2017, 6, e1347742.
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150.
- Sipos, F.; Műzes, G. Cancer Stem Cell Relationship with Pro-Tumoral Inflammatory Microenvironment. Biomedicines 2023, 11, 189.
- Li, C.; Liu, T.; Bazhin, A.V.; Yang, Y. The Sabotaging Role of Myeloid Cells in Anti-Angiogenic Therapy: Coordination of Angiogenesis and Immune Suppression by Hypoxia. J. Cell Physiol. 2017, 232, 2312–2322.
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.P.; Abastado, J.P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162.
- Raber, P.L.; Thevenot, P.; Sierra, R.; Wyczechowska, D.; Halle, D.; Ramirez, M.E.; Ochoa, A.C.; Fletcher, M.; Velasco, C.; Wilk, A.; et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int. J. Cancer 2014, 134, 2853–2864.
- Markowitz, J.; Wang, J.; Vangundy, Z.; You, J.; Yildiz, V.; Yu, L.; Foote, I.P.; Branson, O.E.; Stiff, A.R.; Brooks, T.R.; et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4+ T cells in cancer and measurement of STAT1 nitration. Sci. Rep. 2017, 7, 15424.
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999.
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013, 32, 249–270.
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid. Med. Cell Longev. 2016, 2016, 1580967.
- Rodríguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev. 2008, 222, 180–191.
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77.
- Baniyash, M. TCR zeta-chain downregulation: Curtailing an excessive inflammatory immune response. Nat. Rev. Immunol. 2004, 4, 675–687.
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797.
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440.
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642.
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835.
- Bronte, V.; Kasic, T.; Gri, G.; Gallana, K.; Borsellino, G.; Marigo, I.; Battistini, L.; Iafrate, M.; Prayer-Galetti, T.; Pagano, F.; et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J. Exp. Med. 2005, 201, 1257–1268.
- Lu, T.; Gabrilovich, D.I. Molecular pathways: Tumor-infiltrating myeloid cells and reactive oxygen species in regulation of tumor microenvironment. Clin. Cancer Res. 2012, 18, 4877–4882.
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011.
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res. 2018, 78, 1779–1791.
- Umansky, V.; Shevchenko, I.; Bazhin, A.V.; Utikal, J. Extracellular adenosine metabolism in immune cells in melanoma. Cancer Immunol. Immunother. 2014, 63, 1073–1080.
- Hu, C.E.; Gan, J.; Zhang, R.D.; Cheng, Y.R.; Huang, G.J. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand. J. Gastroenterol. 2011, 46, 156–164.
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249.
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929.
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306.
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194.
- Xu, T.; Jiang, L.; Wang, Z. The progression of HMGB1-induced autophagy in cancer biology. Onco. Targets Ther. 2018, 12, 365–377.
- Alissafi, T.; Hatzioannou, A.; Mintzas, K.; Barouni, R.M.; Banos, A.; Sormendi, S.; Polyzos, A.; Xilouri, M.; Wielockx, B.; Gogas, H.; et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J. Clin. Investig. 2018, 128, 3840–3852.
- Shibata, M.; Nanno, K.; Yoshimori, D.; Nakajima, T.; Takada, M.; Yazawa, T.; Mimura, K.; Inoue, N.; Watanabe, T.; Tachibana, K.; et al. Myeloid-derived suppressor cells: Cancer, autoimmune diseases, and more. Oncotarget 2022, 13, 1273–1285.
- Moliné-Velázquez, V.; Cuervo, H.; Vila-Del Sol, V.; Ortega, M.C.; Clemente, D.; De Castro, F. Myeloid-Derived Suppressor Cells Limit the Inflammation by Promoting T Lymphocyte Apoptosis in the Spinal Cord of a Murine Model of Multiple Sclerosis. Brain Pathol. 2011, 21, 678–691.
- Highfill, S.L.; Rodriguez, P.C.; Zhou, Q.; Goetz, C.A.; Koehn, B.H.; Veenstra, R.; Taylor, P.A.; Panoskaltsis-Mortari, A.; Serody, J.S.; Munn, D.H.; et al. Bone Marrow Myeloid-Derived Suppressor Cells (MDSCs) Inhibit Graft-Versus-Host Disease (GVHD) via an Arginase-1-Dependent Mechanism That is Up-Regulated by Interleukin-13. Blood 2010, 116, 5738–5747.
- Ioannou, M.; Alissafi, T.; Lazaridis, I.; Deraos, G.; Matsoukas, J.; Gravanis, A.; Mastorodemos, V.; Plaitakis, A.; Sharpe, A.; Boumpas, D.; et al. Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J. Immunol. 2012, 188, 1136–1146.
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498.
- Guo, C.; Hu, F.; Yi, H.; Feng, Z.; Li, C.; Shi, L.; Li, Y.; Liu, H.; Yu, X.; Wang, H.; et al. Myeloid-derived suppressor cells have a proinflammatory role in the pathogenesis of autoimmune arthritis. Ann. Rheum. Dis. 2016, 75, 278–285.
- Wang, Z.; Zhu, F.; Wang, J.; Tao, Q.; Xu, X.; Wang, H.; Xiong, S.; Wang, Y.; Zhai, Z. Increased CD14+HLA-DR-/low Myeloid-Derived Suppressor Cells Correlate With Disease Severity in Systemic Lupus Erythematosus Patients in an iNOS-Dependent Manner. Front. Immunol. 2019, 10, 1202.
- Jiao, Z.; Hua, S.; Wang, W.; Wang, H.; Gao, J.; Wang, X. Increased circulating myeloid-derived suppressor cells correlated negatively with Th17 cells in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2013, 42, 85–90.
- Yin, B.; Ma, G.; Yen, C.-Y.; Zhou, Z.; Wang, G.X.; Divino, C.M.; Casares, S.; Chen, S.-H.; Yang, W.-C.; Pan, P.-Y. Myeloid-Derived Suppressor Cells Prevent Type 1 Diabetes in Murine Models. J. Immunol. 2010, 185, 5828–5834.
- Dong, G.; Si, C.; Zhang, Q.; Yan, F.; Li, C.; Zhang, H.; Ma, Q.; Dai, J.; Li, Z.; Shi, H.; et al. Autophagy regulates accumulation and functional activity of granulocytic myeloid-derived suppressor cells via STAT3 signaling in endotoxin shock. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 2796–2807.
- Kotze, L.A.; Leukes, V.N.; Fang, Z.; Lutz, M.B.; Fitzgerald, B.L.; Belisle, J.; Loxton, A.G.; Walzl, G.; du Plessis, N. Evaluation of autophagy mediators in myeloid-derived suppressor cells during human tuberculosis. Cell Immunol. 2021, 369, 104426.
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440.
- Pires-Afonso, Y.; Niclou, S.P.; Michelucci, A. Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 689.
- Ueno, T.; Toi, M.; Saji, H.; Muta, M.; Bando, H.; Kuroi, K.; Koike, M.; Inadera, H.; Matsushima, K. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin. Cancer Res. 2000, 6, 3282–3289.
- Jin, L.; Guo, Y.; Mao, W.; Wang, J.; Jin, L.; Liu, X.; Shou, Q.; Fu, H. Total glucosides of paeony inhibit breast cancer growth by inhibiting TAMs infiltration through NF-κB/CCL2 signaling. Phytomedicine 2022, 104, 154307.
- Shanmugam, G.; Das, S.; Paul, S.; Rakshit, S.; Sarkar, K. Clinical relevance and therapeutic aspects of professional antigen-presenting cells in lung cancer. Med. Oncol. 2022, 39, 237.
- McClellan, J.L.; Davis, J.M.; Steiner, J.L.; Enos, R.T.; Jung, S.H.; Carson, J.A.; Pena, M.M.; Carnevale, K.A.; Berger, F.G.; Murphy, E.A. Linking tumor-associated macrophages, inflammation, and intestinal tumorigenesis: Role of MCP-1. Am. J. Physiol. Liver Physiol. 2012, 303, G1087–95.
- Rhee, I. Diverse macrophages polarization in tumor microenvironment. Arch. Pharm. Res. 2016, 39, 1588–1596.
- Yuen, K.C.; Liu, L.F.; Gupta, V.; Madireddi, S.; Keerthivasan, S.; Li, C.; Rishipathak, D.; Williams, P.; Kadel, E.E., 3rd; Koeppen, H.; et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat. Med. 2020, 26, 693–698.
- Mittal, S.K.; Roche, P.A. Suppression of antigen presentation by IL-10. Curr. Opin. Immunol. 2015, 34, 22–27.
- Luo, X.; Qiu, Y.; Dinesh, P.; Gong, W.; Jiang, L.; Feng, X.; Li, J.; Jiang, Y.; Lei, Y.L.; Chen, Q. The functions of autophagy at the tumour-immune interface. J. Cell Mol. Med. 2021, 25, 2333–2341.
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.U. Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019, 26, 715–727.
- Carrero, J.A.; McCarthy, D.P.; Ferris, S.T.; Wan, X.; Hu, H.; Zinselmeyer, B.H.; Vomund, A.N.; Unanue, E.R. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc. Natl. Acad. Sci. USA 2017, 114, E10418–E10427.
- Jiang, Z.; Jiang, J.X.; Zhang, G.X. Macrophages: A double-edged sword in experimental autoimmune encephalomyelitis. Immunol. Lett. 2014, 160, 17–22.
- Funes, S.C.; Rios, M.; Escobar-Vera, J.; Kalergis, A.M. Implications of macrophage polarization in autoimmunity. Immunology 2018, 154, 186–195.
- Weitz, J.R.; Jacques-Silva, C.; Qadir, M.M.F.; Umland, O.; Pereira, E.; Qureshi, F.; Tamayo, A.; Dominguez-Bendala, J.; Rodriguez-Diaz, R.; Almaça, J.; et al. Secretory Functions of Macrophages in the Human Pancreatic Islet Are Regulated by Endogenous Purinergic Signaling. Diabetes 2020, 69, 1206–1218.
- Wong, P.F.; Wei, W.; Gupta, S.; Smithy, J.W.; Zelterman, D.; Kluger, H.M.; Rimm, D.L. Multiplex quantitative analysis of cancer-associated fibroblasts and immunotherapy outcome in metastatic melanoma. J. Immunother. Cancer 2019, 7, 194.
- Chu, F.; Shi, M.; Zheng, C.; Shen, D.; Zhu, J.; Zheng, X.; Cui, L. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2018, 318, 1–7.
- Li, F.; Yang, Y.; Zhu, X.; Huang, L.; Xu, J. Macrophage Polarization Modulates Development of Systemic Lupus Erythematosus. Cell Physiol. Biochem. 2015, 37, 1279–1288.
- Parsa, R.; Andresen, P.; Gillett, A.; Mia, S.; Zhang, X.M.; Mayans, S.; Holmberg, D.; Harris, R.A. Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes 2012, 61, 2881–2892.
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910.
- Kanayama, M.; Inoue, M.; Danzaki, K.; Hammer, G.; He, Y.W.; Shinohara, M.L. Autophagy enhances NFκB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat. Commun. 2015, 6, 5779.
- Gupta, N.; Jadhav, K.; Shah, V. Emperipolesis, entosis and cell cannibalism: Demystifying the cloud. J. Oral Maxillofac. Pathol. 2017, 21, 92–98.
- Borensztejn, K.; Tyrna, P.; Gaweł, A.M.; Dziuba, I.; Wojcik, C.; Bialy, L.P.; Mlynarczuk-Bialy, I. Classification of Cell-in-Cell Structures: Different Phenomena with Similar Appearance. Cells 2021, 10, 2569.
- Fais, S.; Overholtzer, M. Cell-in-cell phenomena, cannibalism, and autophagy: Is there a relationship? Cell Death Dis. 2018, 9, 95.
- Miao, Q.; Bian, Z.; Tang, R.; Zhang, H.; Wang, Q.; Huang, S.; Xiao, X.; Shen, L.; Qiu, D.; Krawitt, E.L.; et al. Emperipolesis mediated by CD8 T cells is a characteristic histopathologic feature of autoimmune hepatitis. Clin. Rev. Allergy Immunol. 2015, 48, 226–235.
- Chen, L.; Kong, D.; Xia, S.; Wang, F.; Li, Z.; Zhang, F.; Zheng, S. Crosstalk Between Autophagy and Innate Immunity: A Pivotal Role in Hepatic Fibrosis. Front. Pharmacol. 2022, 13, 891069.
- Shi, J.; Zhao, J.; Zhang, X.; Cheng, Y.; Hu, J.; Li, Y.; Zhao, X.; Shang, Q.; Sun, Y.; Tu, B.; et al. Activated hepatic stellate cells impair NK cell anti-fibrosis capacity through a TGF-β-dependent emperipolesis in HBV cirrhotic patients. Sci. Rep. 2017, 7, 44544.
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 2015, 43, 331–342.
- Huang, P.; Wang, F.; Yang, Y.; Lai, W.; Meng, M.; Wu, S.; Peng, H.; Wang, L.; Zhan, R.; Imani, S.; et al. Hematopoietic-Specific Deletion of Foxo1 Promotes NK Cell Specification and Proliferation. Front. Immunol. 2019, 10, 1016.
- Billis, A.; Assis-Mendonça, G.R.; Tavares, T.F.; Parreira, K.; Costa, L.B.E.; Barreto, I.S.; Freitas, L.L.L. Fumarate hydratase-deficient renal cell carcinoma: A tumor with diverse morphology including cannibalism, lymphocytic emperipolesis, and defective autophagy. Ann. Diagn. Pathol. 2022, 56, 151844.
- Florey, O.; Kim, S.E.; Sandoval, C.P.; Haynes, C.M.; Overholtzer, M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 2011, 13, 1335–1343.
- Martins, I.; Raza, S.Q.; Voisin, L.; Dakhli, H.; Law, F.; De Jong, D.; Allouch, A.; Thoreau, M.; Brenner, C.; Deutsch, E.; et al. Entosis: The emerging face of non-cell-autonomous type IV programmed death. Biomed. J. 2017, 40, 133–140.
- Davies, S.P.; Reynolds, G.M.; Wilkinson, A.L.; Li, X.; Rose, R.; Leekha, M.; Liu, Y.S.; Gandhi, R.; Buckroyd, E.; Grove, J.; et al. Hepatocytes Delete Regulatory T Cells by Enclysis, a CD4+ T Cell Engulfment Process. Cell Rep. 2019, 29, 1610–1620.
- Mlynarczuk-Bialy, I.; Dziuba, I.; Sarnecka, A.; Platos, E.; Kowalczyk, M.; Pels, K.K.; Wilczynski, G.M.; Wojcik, C.; Bialy, L.P. Entosis: From Cell Biology to Clinical Cancer Pathology. Cancers 2020, 12, 2481.
- Durgan, J.; Tseng, Y.Y.; Hamann, J.C.; Domart, M.C.; Collinson, L.; Hall, A.; Overholtzer, M.; Florey, O. Mitosis can drive cell cannibalism through entosis. Elife 2017, 6, e27134.
- Kianfar, M.; Balcerak, A.; Chmielarczyk, M.; Tarnowski, L.; Grzybowska, E.A. Cell Death by Entosis: Triggers, Molecular Mechanisms and Clinical Significance. Int. J. Mol. Sci. 2022, 23, 4985.
- Pezzano, M.; Samms, M.; Martinez, M.; Guyden, J. Questionable thymic nurse Cell. Microbiol. Mol. Biol. Rev. 2001, 65, 390–403.
- He, M.F.; Wang, S.; Wang, Y.; Wang, X.N. Modeling cell-in-cell structure into its biological significance. Cell Death Dis. 2013, 4, e630.
- Hendrix, T.M.; Chilukuri, R.V.; Martinez, M.; Olushoga, Z.; Blake, A.; Brohi, M.; Walker, C.; Samms, M.; Guyden, J.C. Thymic nurse cells exhibit epithelial progenitor phenotype and create unique extra-cytoplasmic membrane space for thymocyte selection. Cell Immunol. 2010, 261, 81–92.
- Palm, W.; Park, Y.; Wright, K.; Pavlova, N.N.; Tuveson, D.A.; Thompson, C.B. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 2015, 162, 259–270.
- Krajcovic, M.; Krishna, S.; Akkari, L.; Joyce, J.A.; Overholtzer, M. mTOR regulates phagosome and entotic vacuole fission. Mol. Biol. Cell. 2013, 24, 3736–3745.
- Sun, L.; Meng, Z.; Zhu, Y.; Lu, J.; Li, Z.; Zhao, Q.; Huang, Y.; Jiang, L.; Yao, X. TM9SF4 is a novel factor promoting autophagic flux under amino acid starvation. Cell Death Differ. 2018, 25, 368–379.