Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
HAT, commonly called sleeping sickness, is a zoonotic disease caused by different subspecies of the bloodstream parasite T. brucei and transmitted by the Glossina tsetse fly. Nitroaromatic rings are privileged pharmacophores of many antimicrobials in clinical use, including antifungal and antiparasitic drugs.
- nifurtimox
- drug
- derivatives
1. Nitroimidazoles
Nitroimidazoles are broad-spectrum antimicrobial drugs that have activity against anaerobic Gram-positive and Gram-negative bacteria, parasites and mycobacteria, and are used for the treatment of Helicobacter pylori and Mycobacterium tuberculosis infections [145]. Since new active drugs that effectively shorten the duration of treatment are urgently required to develop effective chemotherapies to combat NTDs, drug repurposing of nitroimidazoles represents a promising source of antitrypanosomal and antileishmanial agents.
1.1. Fexinidazole
The 5-nitroimidazole derivative fexinidazole is also being developed as a potential treatment for other NTDs, as inferred by the ongoing clinical trials supported by the Drugs for Neglected Diseases initiative (DNDi) [67].
Fexinidazole undergoes rapid hepatic metabolization to sulfoxy and sulfone metabolites, which retain in vitro activity against T. cruzi in the low micromolar range. Fexinidazole has been reported to promote massive disorganization of reservosomes, the detachment of the plasma membrane, unpacking of nuclear heterochromatin, mitochondrial swelling, Golgi disruption and alterations in the kinetoplast-mitochondrion complex in T. cruzi [146]. In vivo efficacy after oral administration in a murine model of Chagas disease was confirmed at 50 mg/kg/day (30–40% cure rate) and 100 mg/kg/day (100% cure rate) [147,148]. The antichagasic effect of the final sulfone metabolite was experimentally confirmed in vivo in a chronic infection of T. cruzi, which could be cured with oral fexinidazole sulfone at 100 mg/kg/day for 5 days [149]. Due to the very promising results in preclinical studies with this drug, a phase 2 study of fexinidazole was initiated in 2014 in Cochabamba and Tarija, Bolivia (NCT02498782). Although promising results were obtained, the study had to be interrupted due to safety and tolerability issues. Another phase 2 proof-of-concept study using shorter and lower-dose treatment regimens in adults with chronic Chagas disease (NCT03587766) began in October 2017 in Spain and was completed with final analyses not yet concluded. Results showed that the efficacy of fexinidazole at lower doses and durations was not confirmed after 12 months. Potential use in combination with other drugs or for new indications, such as for immunocompromised patients at risk of reactivation, is being reviewed for the future development of fexinidazole as an antichagasic drug (https://dndi.org/research-development/portfolio/fexinidazole-chagas/; accessed on 3 March 2023).
Fexinidazole and its metabolites were also tested against intracellular amastigotes of L. donovani, yielding a potency similar to miltefosine for the sulfoxy and sulfone forms, but little activity for the parent drug. Assessment of efficacy of fexinidazole in a mouse model of VL revealed that five single daily doses of 200 mg/kg were able to suppress the infection by 98.4%, which was equivalent to that reported for the clinical drugs in use [150]. New World Leishmania species were also susceptible to fexinidazole in vitro and in vivo. Hamster models of CL caused by L. braziliensis and L. guyanensis developed to test the efficacy of fexinidazole showed that this compound became effective from a dose of 200 mg/kg/day, thus resulting in a significant reduction in lesion size. Similarly, a mouse model developed to test the efficacy of oral fexinidazole against L. amazonensis showed a significant effect at 300 mg/kg/day [151]. A phase 2 proof-of-concept trial (NCT01980199) to determine efficacy of fexinidazole at a daily dose of 1800 mg (3 tablets) once a day for 4 days continued by 1200 mg (two tablets) once a day for 6 days in VL patients in Sudan was carried out by DNDi. All patients showed clinical improvement during treatment, and the majority had parasite clearance at the end of treatment. Three patients remained cured until 6 months follow-up. However, the response was not sustained in other patients and relapses were observed. The study was interrupted in 2014 as it failed to show conclusive efficacy in the majority of patients (https://dndi.org/research-development/portfolio/fexinidazole-vl/; accessed on 3 March 2023).
In addition to fexinidazole, there are other nitroimidazole compounds in different stages of development against trypanosomatids (Figure 1).
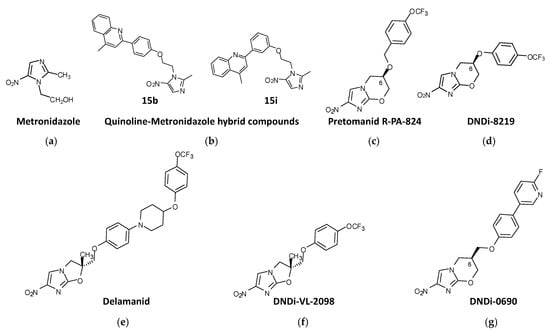
Figure 1. Nitroimidazole compounds in different stages of development against trypanosomatids. (a) Metronidazole. (b) Quinoline-Metronidazole hybrid compounds 15b and 15i. (c) Pretomanid R-PA-824. (d) DNDi-8219. (e) Delamanid. (f) DNDi-VL-2098. (g) DNDi-0690.
1.2. Metronidazole Derivatives
Metronidazole (2-methyl-5-nitroimidazole-1-ethanol) (Figure 1a) was used on patients with Chagas disease, but the results were very inconclusive [152,153]. However, in search of new strategies to reduce the benznidazole administration regimen in Chagas disease, combination therapies with metronidazole were studied. The combination of benznidazole:metronidazole in fixed ratios led to an increase in the potency of benznidazole in vitro. Preclinical studies in infected mice showed that when benznidazole (10 mg/kg) and metronidazole (250 mg/kg) were combined cumulative mortality was less than 40%. Although this combinatorial approach could not reduce the entire parasite load, it protected against cardiac issues induced by experimental T. cruzi infection [154].
Metronidazole has been also administered intralesionally to patients suffering CL as a 0.5% solution for the local treatment of small, non-inflamed and localized lesions. With a success rate of 87%, this treatment was safe, with no serious side effects [155]. However, intralesional injection of 10 mg metronidazole was not as effective as intralesional injection of Pentostam in the treatment of CL in Iraq [156]. Similar results were obtained in a clinical study conducted in Sri Lanka in 2019 with a group of patients with L. donovani CL. Although Pentostam was more effective in healing Leishmania lesions, metronidazole was the most cost-effective drug, curing more than 75% of patients [157].
Quinoline-metronidazole hybrid compounds have been synthesized and tested for antileishmanial activity. Two of these compounds, 15b and 15i (Figure 1b), showed activity against L. donovani promastigotes and amastigotes (>93% parasite killing) at 50 μM, with IC50s = 9.54 and 5.42 μM, respectively, against extracellular promastigotes, and IC50s = 9.81 and 3.75 μM, respectively, against intracellular amastigotes, with no cytotoxicity toward host mammal cells. In vivo studies were carried out with a mouse model of L. donovani infection with these compounds, which were administered intraperitoneally for 5 consecutive days in a dose range of 12.5–200 mg/kg daily. The results showed that both compounds were able to inhibit parasitic load in the organs of infected mice, with compound 15i exhibiting better efficacy than 15b, and inhibit parasite burden in the livers and spleens (>80%) of infected mice treated with a dose of 50 mg/kg/day for 5 consecutive days [158].
1.3. Nitroimidazo-Oxazines and Nitroimidazo-Oxazoles
The 4-nitroimidazo-oxazine pretomanid (PA-824), (S)-2-nitro-6-[4-(trifluoromethoxy) benzyloxy]-6,7-dihydro-5H-imidazo [2,1-b][1,3]oxazine) (Figure 1c), which was approved in 2019 by the FDA to treat highly challenging cases of tuberculosis [159], also exhibited antiparasitic activity against trypanosomes and leishmanias. PA-824 consists of a mix of stereoisomers R and S with different biological activities in vitro. The R enantiomer was shown to be five times more active than the S enantiomer against all forms of the parasite. Treatment with 100 mg/kg with the R-stereoisomer of PA-824 suppressed infection by 99.9% and effectively cured the murine model of L. donovani VL in contrast to the S-stereoisomer, which was no more than 35% effective with the same dosification [160]. These results were also replicated in vitro against mammalian stages of T. brucei and T. cruzi, where the S-stereoisomer was largely inactive, in contrast to the R-stereoisomer, which showed higher trypanocidal activity [160].
PA-824 has served as a chemical scaffold for many other compounds with antitrypanosomal activity. From a library of ∼900 pretomanid analogs, compound DNDI-8219 ((6R)-2-Nitro-6-[4-(trifluoromethoxy)phenoxy]-6,7-dihydro-5H-imidazo [2,1-b][1,3]oxazine)) (Figure 1d) was identified. This compound showed strong (submicromolar) antileishmanial in vitro activity against both L. infantum and L. donovani intramacrophage amastigotes, with broad-spectrum activity against a range of reference strains and clinical isolates of Leishmania. In a mouse model of chronic VL, this compound gave rise to ≥97% inhibition when administered at 25 mg/kg bid for 5 days [161,162]. DNDI-8219 has shown interesting antichagasic activity against a chronic infection of T. cruzi mouse model when administered at 50 mg/kg/day for 5 consecutive days [163].
Delamanid (OPC-67683) is a nitro-dihydro-imidazooxazole (Figure 1e) resembling PA-824 that received conditional approval by the EMA for the treatment of multidrug-resistant tuberculosis [164]. Like PA-824, delamanid is a racemic mixture of two steroisomers, the R enantiomer showing better in vitro activity (submicromolar order) against promastigotes, axenic amastigotes and intracellular amastigotes of L. donovani than the S enantiomer. Both enantiomers provided good safety results in mammalian cells [165]. The same study tested the in vivo efficacy of an oral formulation of delamanid at different doses in a murine model of L. donovani, showing that either 30 or 50 mg/kg/day orally administered twice daily for 5 consecutive days compared favorably with the standard miltefosine regimen [165].
From the in vitro evaluation of a library of 72 nitroimidazo-oxazoles against L. donovani intracellular amastigotes, 25 compounds with IC50 values ≤0.1 μM were identified. Among them, the racemate DNDI-VL-2001 (2-methyl-6-nitro-2-[[4-(trifluoromethoxy)phenoxy] methyl]-2,3dihydroimidazo [2,1-b]oxazole), a 6-nitroimidazo-oxazole derivative, was able to reduce by 99% the parasite load in the liver of a mouse model of L. donovani VL at a dose of 25 mg/kg/day for 5 consecutive days via the oral route. R and S enantiomers were tested orally in a mouse model with five dose regimens ranging from 0.78 to 12.5 mg/kg. The R enantiomer DNDI-VL-2098 (Figure 1f) was identified as the most potent and safe stereoisomer. In addition, in a hamster model of VL, DNDI-VL-2098 promoted T-cell differentiation towards a Th1 type response and induced the production of extracellular nitric oxide [166]. Further QSAR studies of this compound were conducted in order to improve aqueous solubility and other drug-like features without compromising in vivo efficacy [167]. However, DNDI-VL-2098 did not continue to further studies due to safety issues (https://dndi.org/research-development/portfolio/vl-2098/; accessed on 3 March 2023).
The 7-substituted nitroimidazooxazine analogue of DNDI-VL-2098, DNDI-0690 (Figure 1g) was also identified from a library of 70 nitroimidazole-derived compounds by DNDi through an agreement with the TB Alliance. In mice, DNDI-0690 is rapidly absorbed into the bloodstream, showing a gastrointestinal absorption delay of 2.5 h before reaching peak plasma concentration and an estimated elimination half-life of >4 h [168]. The strong antileishmanial effect of this compound was demonstrated against intramacrophage amastigotes of L. donovani and L. infantum in vitro and in a hamster model of L. infantum VL in vivo, where it produced >99% inhibition of parasite load when administered orally at a dose of 12.5 mg/kg bid for 5 days [169]. In addition to its activity against VL, DNDI-0690 demonstrated excellent in vitro activity against three Old World and three New World strains of cutaneous Leishmania (EC50 < 5 μM). In a mouse model of L. major CL, DNDI-0690 exhibited > 95% efficacy at a dose of 50 mg/kg, while topical solutions applied directly to the skin lesion were < 50% active [129]. In another study, when DNDI-0690 was administered orally at a dose of 50 mg/kg daily for 10 consecutive days to two mouse models of L. major and L. mexicana CL in vivo, complete parasitological cure occurred [168]. Film-forming systems for delivery have been developed to increase the effectiveness of DNDI-0690 for the topical treatment of CL. In a L. major mouse model of CL, only the Eudragit RS formulation (containing butyl sebacate) resulted in a reduced parasite load, but without reduction in lesion size, which suggested that inadequate amounts of DNDI-0690 reached the parasites in the dermal layers of the skin upon topical application [170]. A multiple ascending dose phase 1 study of DNDI-0690 run in early 2020 in healthy adults demonstrated a favorable safety profile for single-dose administration (NCT03929016).
Recently, a second generation of DNDi-0690 analogues with increased solubility and safety led to the pyridine derivate (7-Methyl-2-nitro-7-({[5-(trifluoromethyl)pyridin-2-yl]oxy}methyl)-6,7-dihydro-5H-imidazo [2,1-b][1,3]oxazine) (Figure 1h). It exhibited increased bioavailability in mice and similar efficacy (reduction in the parasite burden >96%) in a model of VL when administered orally at a dose of 50 mg/kg bid for five days to mice infected with L. donovani [171].
2. Nitrofurans
Nitrofurans (Schiff base derivatives of 5-nitrofuraldehyde) are broad-spectrum redox-active antibiotics with dose-dependent bacteriostatic or bactericidal activity [172,173]. These compounds have been used in animal feeds, pharmaceuticals and other applications [174]. The hydrazine moiety of these compounds aids in the overall chemical stability of the nitrofuran ring due to its zwitterionic properties [175,176] and is responsible for the antipathogenic activity of nitrofurans [174].
There are several nitrofuran compounds in different stages of development against trypanosomatids (Figure 2).
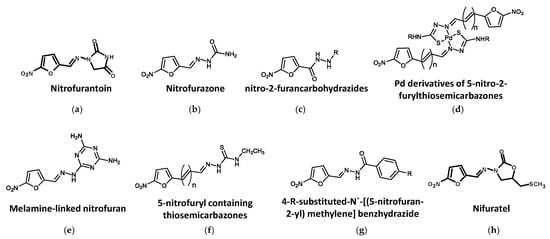
Figure 2. Nitrofuran compounds in different stages of development against trypanosomatids. (a) Nitrofurantoin. (b) Nitrofurazone. (c) 5-nitro-2-furancarbohydrazides. (d) Pd derivatives of 5-nitro-2-furylthiosemicarbazones. (e) Melamine-linked nitrofuran. (f) 5-nitrofuryl containing thiosemicarbazones. (g) 4-R-substituted-N′-[(5-nitrofuran-2-yl) methylene] benzhydrazide. (h) Nifuratel.
2.1. Nitrofurantoin
Nitrofurantoin (1-[[[5-nitro-2-furanyl]methylene]amino]-2,4-imidazolidinedione) (Figure 2a) is used for the treatment of human bacterial urinary tract infections [177] and has been listed in the WHO model list of essential medicines [87]. Early studies reported the in vitro antiparasitic activity of nitrofurantoin against certain strains of T. cruzi [178]. Nitrofurantoin and other N-alkyl and benzyl-substituted analogs were recently tested for their trypanocidal activity, showing strong trypanocidal activity (submicromolar range) against T. congolense and T. b. rhodesiense and low cytotoxicity. However, only nitrofurantoin had significant trypanocidal activity when it was administered orally at a dose of 10 mg/kg daily for 7 consecutive days to mice infected with T. congolense IL3000 strain [179]. In an ascending dose experiment with nitrofurantoin administered orally to mice infected with T. congolense, doses of 30 to 100 mg/kg for 7 consecutive days were safe and curative and resulted in the complete healing of the animals [180], thus pointing to nitrofurantoin as a potential scaffold for the synthesis of new compounds with antitrypanosomal activity.
2.2. Nitrofurazone
Nitrofurazone (5-nitro-2-furaldehyde-semicarbazone), a hydrazinecarboxamide containing 5-nitrofuran, lacks the second cyclic substituent in its structure (Figure 2b). Formulated as a topical cream, it is used for the treatment of superficial wounds, second- and third-degree burns, ulcers and skin infections [181,182]. The trypanocidal activity of nitrofurazone was earlier demonstrated on the intracellular forms of T. cruzi in experimental Chagas disease [183]. More recently, a novel nitrofurazone derivative, hydroxymethylnitrofurazone, together with nitrofurazone, was tested in cell cultures (LLC-MK2) infected with amastigote and trypomastigote forms of T. cruzi. The nitrofurazone derivative exhibited better trypanocide activity and lower mutagenicity potential than nitrofurazone, with 100% inhibitory activity against amastigotes and trypomastigotes at 5 μM [184].
2.3. Other Nitrofuran Derivatives
It has been reported that several 5-nitro-2-furancarbohydrazines (Figure 2c) demonstrated good activity against the intracellular amastigote stage of both T. cruzi and L. infantum and T. brucei bloodstream trypomastigotes [185]. Some compounds derived from palladium nitrofurylthiosemicarbazone ([PdCl2(HL)] and [Pd(L)2]) (Figure 2d), which are trypanothione reductase inhibitors, were 1.7-fold more active than nifurtimox against T. cruzi epimastigotes [186].
The melamine derivate of nitrofuran WSP934 (Figure 2e) was reported to have very potent activity against T. b. rhodesiense (IC50 about 11 nM). In addition, when 20 mg/kg of this compound was administered daily by the intraperitoneal route for 4 consecutive days to mice infected with T. b. brucei, a curative effect was observed [187,188]. In another study [189], when the dose was increased to 40 mg/kg/day, mice infected with T. b. rhodesiense were also cured. These authors also observed that oral treatment with 100 mg/kg for 4 days gave rise to moderate activity, whereas 10 doses of 50 mg/kg given intraperitoneally did not provide cure in a stage 2 rodent model of infection.
5-Nitrofuryl derivatives containing thiosemicarbazones (Figure 2f) have been reported to possess high anti-T. cruzi activity. In vitro activity against T. cruzi of some of these molecules (6, 8, 10 and 12), which are 3-(5-nitrofuryl) acroleinyl derivatives containing H, methyl, ethyl and phenyl moieties, respectively, was superior to that of nifurtimox [190]. In another study, 19 compounds containing a common 5-nitrofuran core with different substituent groups at the 2-position on the furan ring (thiosemicarbazones, semicarbazones and carbazates) were tested in vitro for their activity against the bloodstream-form of T. brucei. From the 19 compounds, 13 had IC50s <1 μM, with the thiosemicarbazones HC1, HC2 and HC4, the carbazates HC10 and HC11, and the semicarbazone nitrofurazone providing values of <250 nM. The carbazate HC10 was the most potent agent against both bloodstream forms of T. brucei and mammalian cells, with a promising selectivity index of 116 [191].
A series of nitroheterocyclic compounds (BSF series) were tested as new alternatives against L. infantum [192]. Five compounds, 4-R-substituted-N′-[(5-nitrofuran-2-yl) methylene] benzhydrazide (R = H, Cl, NO2, CH3, C4H9) (Figure 2g), were obtained through the molecular modification of nifuroxazide, 4-hydroxy-N′-[(5-nitrofuran-2-yl)methylene] benzohydrazide and provided activity against this parasite in a dose-dependent fashion. BSF-H, BSF-Cl and BSFNO2 provided the best IC50 values in promastigotes (0.76 μM, 0.72 μM and 0.58 μM, respectively). The authors also showed that BSF-H and BSF-Cl were effective in infected macrophages, too, thereby concluding that these nitroheterocyclic compounds were promising antileishmanial drugs.
In a recent published screening of two commercial collections of 1,769 repositioning drugs using splenic explants from mice infected with an infrared fluorescent L. donovani strain [193], 43 compounds with leishmanicidal activity > 1μM were selected. Among the selected compounds, several with heterocyclic structures were identified as the most potent, including nifurtimox, nitrofurantoin, nitrofurazone, PA-824, fexinidazole and the fungicide Nifuratel (Macmiror) (Figure 2h), the latter being a synthetic nitrofurantoin derivate active against most agents causing genital urinary infections [194], whose potency and selectivity indicate it as a promising antileishmanial drug [143].
Further studies in a mouse model of chronic VL with a dose of nifuratel administered orally at 50 mg/kg bid for 10 consecutive days resulted in a reduction in parasite load of more than 80%. In addition, intralesional administration of nifuratel in a model of L. major CL resulted in parasitological cure, making this compound a potential candidate for the treatment of this insidious form of the disease [143]. Nifuratel was also tested in combination with miltefosine, and the 1/30 ratio (nifuratel/miltefosine) exhibited synergic antileishmanial effect both in vitro in axenic amastigotes of L. donovani and ex vivo in intramacrophagic forms. Administration of 50 mg/kg nifuratel (twice daily) in combination with 10 mg/kg miltefosine (once daily) for 10 days produced in a mouse model of VL a significant reduction in total parasite burden, especially in the thymus, liver and bone marrow, although it was not able to completely clear the presence of parasites in the spleen [195].
3. Other Nitroheterocycles
Other active nitroheterocyclic compounds have been tested against trypanosomatids (Figure 3).
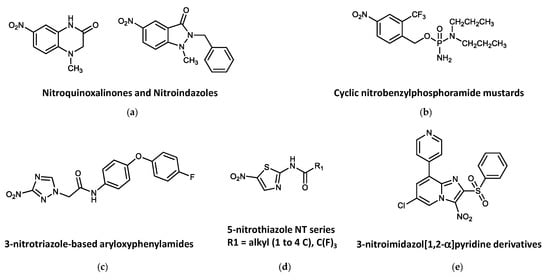
Figure 3. Other active nitroheterocyclic compounds tested against trypanosomatids. (a) Nitroquinoxalinones and Nitroindazoles. (b) Cyclic nitrobenzylphosphoramide mustards. (c) 3-nitrotriazole-based aryloxyphenylamides. (d) 5-nitrothiazole NT series. (e) 3-nitroimidazol [1,2-α] pyridine derivatives.
From an in-house library comprising 76 nitroheterocycles and related compounds tested in vitro against T. b. rhodesiense, six hits were identified showing interesting activity (IC50 ≤ 10 μM) and fair selectivity (SI > 17). The best antitrypanosomal compounds were those with the quinoxalin-2-one (Figure 3a) scaffold (IC50 values <15 μM and SI values in the range of 3.6–39.7). Two compounds, which derived from the 7-nitroquinoxalin-2-one and 5-nitroindazole scaffolds, respectively, were found to be of particular interest because of their established oral bioavailability in mice [196].
A library of nitrobenzylphosphoramide mustards (Figure 3b) was assessed in vitro against L. major. Five compounds showed significant activities (IC50 values of <10 μM) against both parasite forms of L. major, showing no toxicity to the mammalian cells at concentrations up to 100 μM. Three of these compounds contained halogen substituents on the nitrobenzyl ring and were identified as being preferred substrates for the L. major type I NTR and provided IC50 values ≤ 1.09 μM against the intracellular stage and SIs > 92 [197].
3-Nitro-1H-1,2,4-triazole- and 2-nitro-1H-imidazole-based amides with aryloxy-phenyl cores (Figure 3c) demonstrated significant activity against T. cruzi amastigotes. The 3-nitrotriazole-based derivatives showed high potency as anti-T. cruzi agents at subnanomolar concentrations with high selectivity. Antitrypanocidal activity of the 2-nitroimidazole analogs was only moderate and poorly selective. Interestingly, both types of compounds were also active against L. donovani axenic amastigotes, with excellent selectivity for the parasite. Despite the activity against L. donovani axenic amastigotes, 3-nitrotriazole-based analogs were not particularly active in infected macrophages, whereas three 2-nitroimidazole-based analogs were also moderately active against infected macrophages. On the contrary, these compounds could not demonstrate selective activity against T. b. rhodesiense. Mice infected with T. cruzi were able to reduce the parasite load to undetectable limits after a 5-day intraperitoneal treatment with 13 mg/kg/day of three 3-nitrotriazole-based aryloxyphenylamides. These compounds constitute a promising generation of antitrypanosomal agents [198].
Fifteen compounds based on a 2-amide 5-nitrothiazole structure (Figure 3d) were tested for growth-inhibitory activity against the bloodstream form of T. brucei. Only compounds NT2, NT4, NT6, NT7 and NT11 provided appreciable trypanocidal activity (IC50s < 10.0 μM), with no growth-inhibitory effects at concentrations of up to 100 μM in mammalian cells [199].
Several 3-nitroimidazo [1,2-α]pyridine derivatives (Figure 3e) were synthesized and evaluated for their in vitro antiparasitic activity in both Leishmania spp. and T. b. brucei. Although the biological activity of these compounds against Leishmania was poor, they inhibited bloodstream forms of T. b. brucei in the submicromolar range, with excellent selectivity index values (≥2500) in mammalian cell cultures. The best antitrypanosomal molecule in this series (compound 8: 8-bromo-6-chloro-2-(methylsulfonylmethyl)-3-nitroimidazo [1,2-a]pyridine) showed an EC50 = 17 nM and an SI = 2650, was not genotoxic, displayed improved aqueous solubility and better in vitro pharmacokinetic properties, and was well tolerated by mice after repeated oral administrations of 100 mg/kg for 5 days [200].
This entry is adapted from the peer-reviewed paper 10.3390/biom13040637
This entry is offline, you can click here to edit this entry!