Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Multiple myeloma (MM) is the second most prevalent hematologic malignancy. One of the significant obstacles in treating most MM patients is drug resistance, especially for individuals who have experienced relapses or developed resistance to such cutting-edge treatments. One of the critical processes in developing drug resistance in MM is autophagic activity, an intracellular self-digestive process. In multiple myeloma, it has been shown that High mobility group box protein 1 (HMGB1)-dependent autophagy can contribute to drug resistance.
- autophagy
- drug resistance
- multiple myeloma
1. Introduction
Autophagy plays an essential role in the digestion and degradation of accumulated excess proteins and impaired or damaged organelles via a lysosome-dependent pathway [1]. Activation of autophagy during stress conditions such as starvation or production of reactive oxygen species (ROS) provides a source of energy and removes excess components, leading to cellular homeostasis and survival [2]. Nevertheless, during certain conditions, extensive autophagic activity may lead to type 2 programmed cell death (PCD) [3].
Three main types of autophagy that have been identified include microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy, hereafter referred to as “autophagy”. The non-selective lysosomal degradative process that engulfs cellular components is known as microautophagy [4]. Direct membrane invagination forms vesicles [5] which, in turn, carry cell ingredients to the lysosomal vesicle, starting the breakdown of soluble cytoplasmic components or other completely integrated organelles, such as peroxisomes (Figure 1a). Proteostasis maintenance along with the cellular response to unfavorable situations are both facilitated by CMA [6]. In CMA, selective identification of substrates occurs in the absence of vesicles, followed by transfer through cytoplasmic hsc70/co-chaperones to the lysosomal membrane [7]. At the lysosomal surface, substrate internalization occurs by a membrane transfer compound that is created through multimerization of the CMA substrate–chaperone receptor, known as lysosome-associated membrane protein type 2A (LAMP-2A) [8].
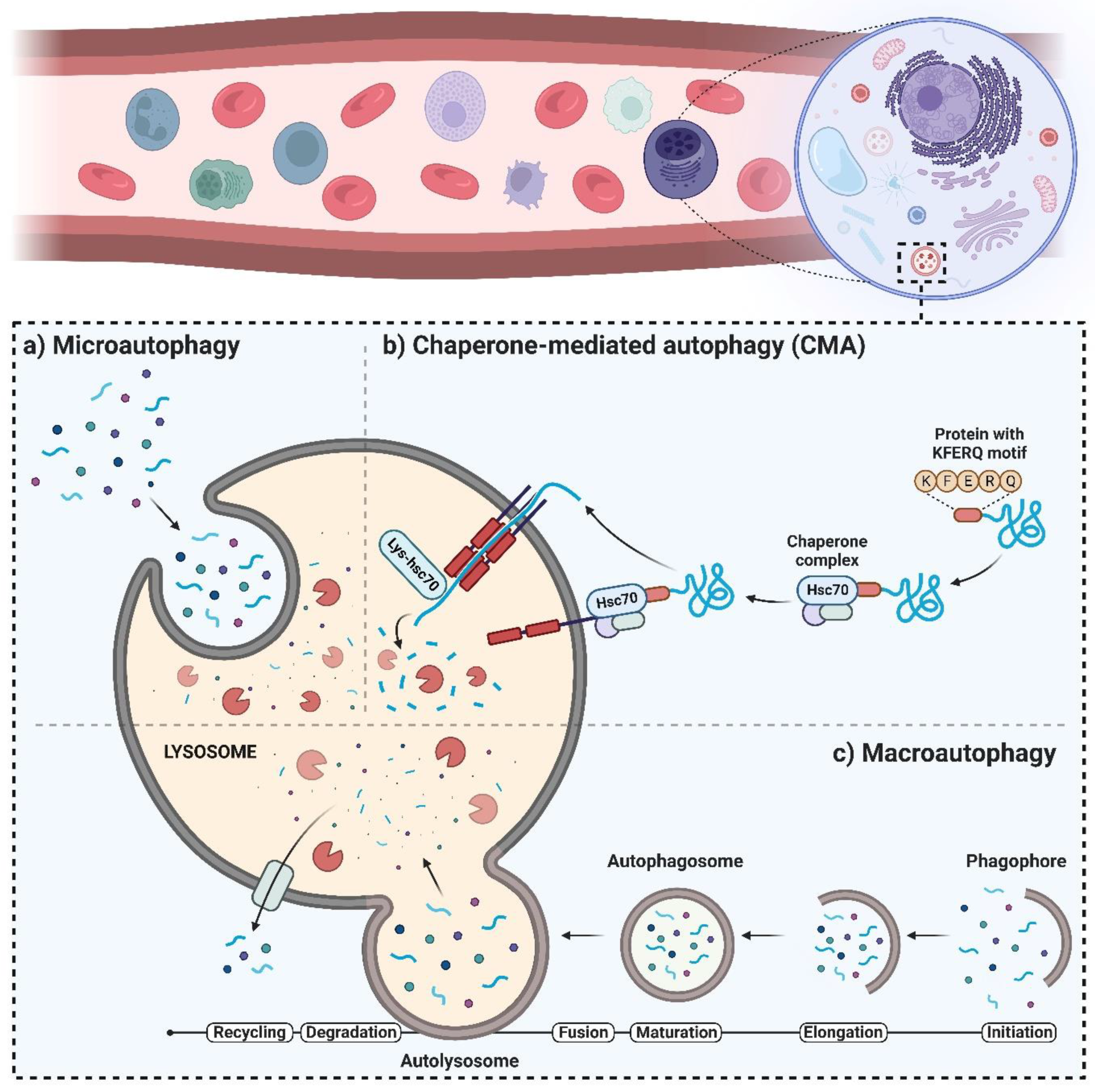
Figure 1. Different types of autophagy: (a) microautophagy, (b) chaperone-mediated autophagy (CMA), and (c) macroautophagy.
The multimerization step leads to the translocation and degradation of the substrate (Figure 1b). During the normal state, very low levels of CMA exist in most cells [9], while stress factors such as starvation and oxidative stress upregulate CMA and enhance the number of lysosomes with active CMA that contain CMA substrates and hsc70 [10]. In research conducted by Nikesitch et al. [11], CMA was shown to increase in bortezomib-resistant MM, and its inhibition made bortezomib-resistant cells more susceptible. Moreover, there was a significant increase in the protein levels of LAMP2A, the rate-limiting component of the CMA pathway, in both MM patients who were resistant to bortezomib as well as in the bortezomib-resistant cell line model. Furthermore, compared to the parent cell line that was bortezomib-sensitive, the bortezomib-resistant cells had increased baseline CMA activities. Bortezomib-resistant cells became susceptible to bortezomib when CMA was inhibited, and the in vitro combination of bortezomib with CMA inhibition was more cytotoxic to myeloma cells than bortezomib alone. The findings of this research reveal that the elevation of CMA is a potential bortezomib-resistance mechanism and a new target for treating bortezomib-resistant MM. Autophagy is a highly conserved process regulated through various multistage signaling processes [12] to ensure the recycling of cytosolic organelles, proteins, macromolecules, and invasive microorganisms. During the autophagy process, an expanding bilayer structure called “autophagosome” isolates the substrates to be broken down in the lysosomes [13] (Figure 1c). Afterward, the degraded and digested components are transferred into the cytoplasm [14] to supply an alternate source of energy and maintain cellular homeostasis. During normal conditions, autophagy is activated to a small extent in most human cells to sustain homeostasis and/or to enhance cell viability in response to unfavorable situations [15]. Disruption of the regulatory mechanisms and/or mutations of autophagy genes triggers the occurrence and/or development of various human diseases and disorders, such as cancer.
2. The Autophagy Machinery and Its Regulatory Mechanisms
Several central markers interact during autophagic activity, among which are a group of genes known as AuTophaGy-related genes (ATGs). Thirty ATGs have been discovered so far in the yeast Saccharomyces cerevisiae [16], and their analogs have been identified in other eukaryotes. Constant expression of ATGs is required to regulate autophagy. The role and importance of Atg proteins in the formation of autophagosomes have been well investigated within different stages of autophagy [13][17]. As shown in Figure 1c, the autophagy process consists of multiple sequential steps, including initiation, elongation, maturation, fusion, and degradation. The mammalian target of rapamycin (mTOR) is considered the master regulator of autophagy [18][19] and, in eukaryotes, is a highly conserved protein kinase [20]. Upon interaction of mTOR with multiple proteins, two distinct complexes form—mTOR complex 1 (mTORC1) and 2 (mTORC2)—which have different reactivity to rapamycin, as well as different downstream outputs and upstream inputs [21][22]. Through the inhibition of catabolic activities (such as degradation of mRNA and activation of autophagy) on one hand, and by the activation of anabolic procedures (such as ribosome biogenesis, transcription, and protein synthesis) on the other hand, mTOR is involved in controlling cell growth [23]. Autophagy inducers, such as lack of oxygen, ROS, DNA damage, and starvation, negatively regulate the activation of mTOR [24] (Figure 2). In particular, mTORC1 is sensitive to starvation, which inactivates mTOR [25], leading to autophagy induction. Under stress situations, phosphorylation of Atg13 by mTOR does not occur, which results in the binding of Atg13 to Unc-51-like kinase-1/2 (ULK1/2) that then triggers the enzymatic capability to induce the formation of the phagophore [26][27]. Next, to complete the phagophore formation step, ULK1/2–Atg13 uses a kinase network [28] that includes UV radiation resistance-associated gene protein (UVRAG), vacuolar protein sorting-34 (Vps34), phosphatidylinositol-3-kinase class II (PI3KC3), Beclin-1, and autophagy and Beclin-1 regulator-1 (AMBRA-1) [29]. Beclin-1, a Bcl-2-homology (BH)-3 domain protein, is the mammalian orthologue of yeast Atg6 [30] and, depending on the type of the proteins (inducers or inhibitors) that bind to Beclin-1, functions as a central regulator to induce or inhibit autophagic activity [31] (Figure 2).
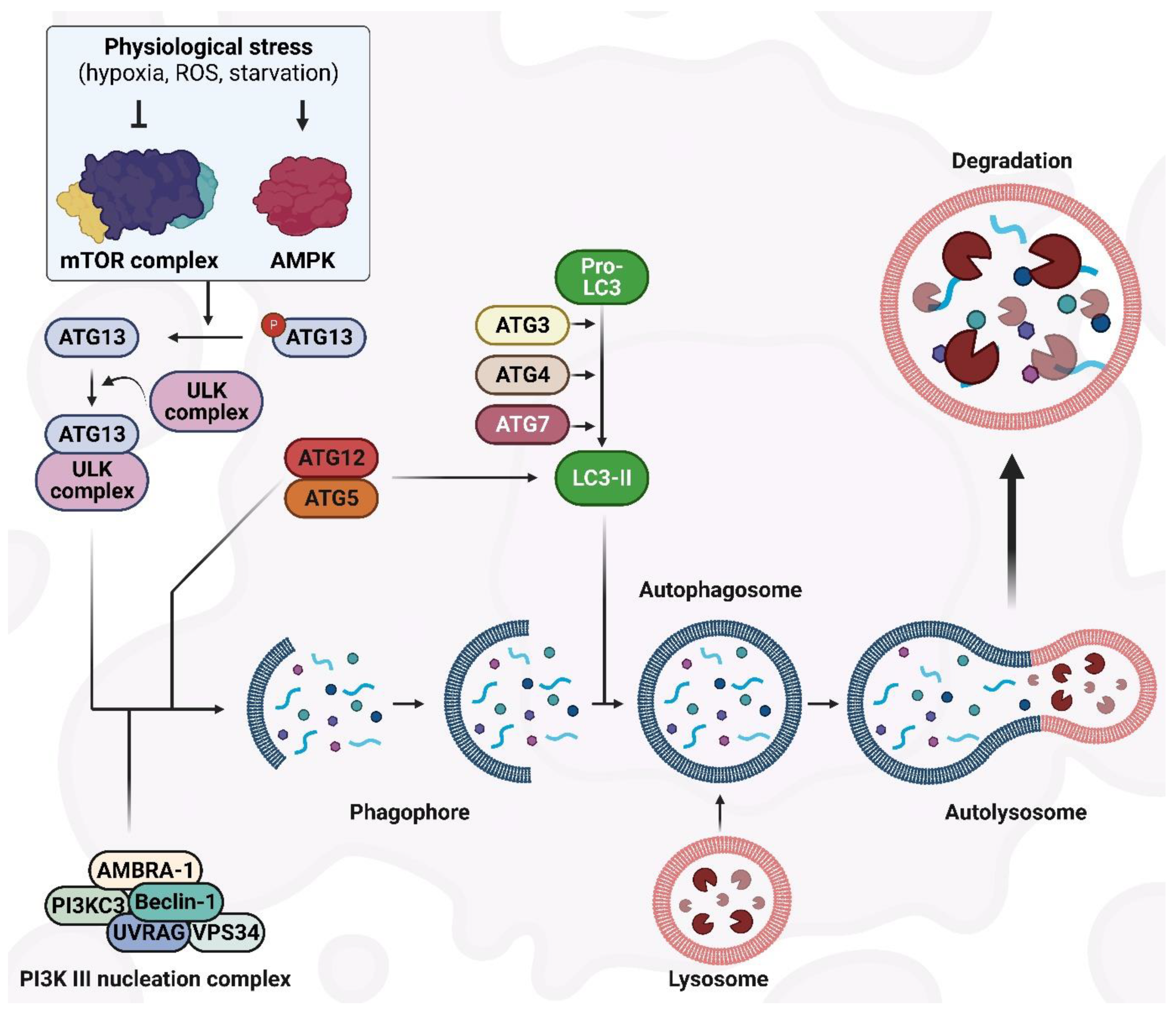
Figure 2. The autophagy machinery and its regulatory mechanisms.
Phagophore elongation continues with two ubiquitin-like systems: the substrate receptor p62/sequestome1 (p62/SQSTM1) and the light chain3 (LC3) systems. Full-length LC3 can be found in the cytosol, and its activity depends on the processing of microtubule-associated proteins. Following autophagy stimulation, Atg4, a cysteine protease, proteolytically cleaves pro-LC3 to generate the soluble cytoplasmic form, LC3-I. Atg7, an E1-homologous factor, activates Atg4 with ATP to induce LC3-I movement to Atg3, an E2-homologous transfer factor [12]. Thereafter, active LC3-I conjugates with phosphatidylethanolamine (PE) to produce the lapidated form, LC3-II [32]. LC3-II can be detected on either the internal or external autophagosome membrane, and the interactions between Atg5–Atg12 determine its recruitment and combination. LC3-II is involved in the regulation of autophagosome–lysosome membrane fusion and the selection of cargo for digestion purposes [33]. p62/SQSTM1 is considered to be an autophagy marker and, through the LC3-connecting site, associates the protein ubiquitin-binding region to LC3-II to promote the recycling of ubiquitinated proteins at the autolysosome surface [34] (Figure 2).
Regulation of the autophagy process occurs in a variety of ways [35]. mTOR is the main regulator that functions like a signaling regulation site downstream of the insulin signaling, ATP content, and growth factor receptors. After starvation and reduced ATP levels, mTOR is suppressed and leads to the activation of adenosine 5′-monophosphate–activated protein kinase (AMPK) [36]. Moreover, inhibition of mTOR under the AMPK effect results in the activation of autophagy in a hypoxia-inducible factor (HIF)-independent and -dependent manner [37]. The endoplasmic reticulum (ER) stress response is triggered in the presence of hypoxia and through the unfolded protein response, which attenuates the mitochondrial mass along with the mitochondrial function in oxidative phosphorylation while enhancing autophagic activity to eliminate the ER-compacted portions [38]. This adaptation to hypoxia restrains the wasteful consumption of ATP by ER and limits the generation of ROS in the mitochondria. In addition, promoted autophagy can produce ATP from catabolism once there is limited ATP production by oxidative phosphorylation [39].
3. Significance of Autophagic Activity in the Survival of MM Cells
In tumor cells, metabolic stress triggers autophagy as an alternative source of energy and metabolites [40], promoting an adaptive cellular response to cancer treatments [41]. Autophagy is crucial in alleviating drug-induced cell death through chemoresistance in hematologic malignancies [42]. MM is a hematologic malignancy identified by the proliferation of monoclonal plasma cells in the bone marrow (BM). After non-Hodgkin’s lymphoma, MM is the second-most prevalent hematologic cancer (representing 10% of all hematologic cancers), substantially increasing in incident cases worldwide over the previous 25 years [43][44]. MM ontogeny is distinguished by various stages. The initial stage, defined as monoclonal gammopathy of unknown significance (MGUS), has no obvious signs, a low level of plasma cell (PC) replication, and low immunoglobulin production [45][46]. Individuals with MGUS have a 1% annual risk of developing MM by age 20 [47]. In some instances, it is feasible to diagnose a middle phase known as smoldering multiple myeloma (SMM), which has a larger immunoglobulin (Ig) production yet remains asymptomatic [48]. Once patients start to show symptoms, the disorder is known as MM, which may develop as an intramedullary or extramedullary condition [49][50]. The extramedullary condition is correlated with the worst outcome, particularly in the spreading phase of plasma cell leukemia, where high numbers of malignant PCs in peripheral blood circulation are detectable [51][52]. Monoclonal immunoglobulins (Igs), which are produced in large quantities by MM cells, cause potentially harmful misfolded or unfolded proteins to localize on the ER. Due to the increased proliferation capacity and Ig production, autophagy plays a critical role in the survival of MM cells to digest the extra protein aggregates [53]. Autophagy inhibition through the knock down of Beclin-1 expression or incubation in the presence of autophagy inhibitors, such as 3-methyladenine (3-MA) and chloroquine, results in the apoptosis of MM cells [54][55] and prevents autophagosome formation. Recently, Wang et al. [56] showed that elaiophylin, a potent inhibitor of the late autophagy phase, exhibits anti-MM cell activity through the inhibition of autophagic flux and sustained activation of ER stress-mediated apoptosis.
Moreover, basal autophagy is tightly controlled to avoid autophagic cell death. Lamy et al. [57] point to the heterodimeric protease caspase-10/FLIPL as a pro-survival factor that restricts basal autophagy via cleavage of the Bcl-2-interacting protein Bcl-2-associated transcription factor 1. Inhibition or silencing of caspase-10 stabilizes Bcl-2-associated transcription factor 1, which displaces Bcl-2 from Beclin-1, resulting in excessive autophagy and consequent MM cell death. Autophagy has been described in the literature as an MM pro-survival strategy that can provide a preventive impact during drug therapy, as drug-resistant MM cells are able to tolerate the cytotoxic effects of drugs through autophagy [58]. Bortezomib and carfilzomib are two proteasome inhibitors that are used as the primary agents for individuals with recently diagnosed or relapsed MM. Although these drugs exert an anti-cancer impact initially, patients frequently develop resistance. MM cells that resist bortezomib generate more autophagosomes and have more AMPK content than cells that are susceptible to the drug.
A decreased activity of AMPK impairs the generation of autophagosomes [59]. Riz et al. [60] showed that Kruppel-like factor 4, a transcription factor, binds to the SQSTM1 gene promoter region and makes MM cells resistant to carfilzomib. According to Hoang et al. [61], exposing MM cells to inhibitors of mTOR and promoters of ER stress increases autophagy even more. mTOR is a famous autophagy inhibitor and treating MM cells with the drug inhibitor 3-MA results in a dose-dependent induction of autophagic cell death. Exposing MM cells to bortezomib along with autophagy inhibition simultaneously lead to synergistic toxic effects [61]. Suppression of the PI3K/Akt/mTOR signaling pathway positively correlates with the activation of autophagy under ER stress [62]. Fu et al. [63] showed that ER stress enhances autophagic activity and apoptosis while decreasing cell expansion via suppression of the PI3K/Akt/mTOR pathway in a large cohort of MM individuals classified as susceptible and resistant patients depending on the effectiveness of the chemotherapeutic approach. In addition, ER stress may return DR through the PI3K/Akt/mTOR axis. Inside the cells, nicotinamide adenine nucleotide is critical in regulating various cellular processes. It is highly expressed in MM cells and is involved in drug resistance as well as cell growth [64]. Cea et al. showed that suppression of nicotinamide phosphoribosyltransferase (NAMPT), a rate-limiting enzyme engaged in nicotinamide adenine nucleotide production, evoked related cytotoxic effects against MM cells resistant to routine anti-MM drugs in vitro and in vivo, and inhibits the preventive properties of IGF-1, interleukin-6, and BM stromal cells. The cytotoxicity of the NAMPT inhibitor FK866 is caused by autophagy activation via inhibition of the mTORC1/Akt and ERK1/2 pathways [64]. In cancers, autophagy plays dual roles. Numerous cancers, including pancreatic, hepatic, and colorectal cancers, along with hematologic malignancies such as lymphoma, leukemia, and myeloma have been linked to impaired autophagic activity.
Additionally, changes in autophagic activity can lead to the development of drug resistance upon undergoing chemotherapy with drugs such as doxorubicin, etoposide and cisplatin [65]. Nevertheless, as autophagy has both tumorigenic and tumor-suppressive impacts, its significance in cancer is not precisely known [4][40]. In tumors, autophagy-related genes are frequently absent. For example, Beclin1, a gene that produces a crucial protein member of the PI3K complex, is typically downregulated in human breast cancer and carries monoallelic deletions [66]. Moreover, it has been proposed that Beclin1 and its positive activator, UV radiation resistance-associated gene (UVRAG), are crucial in activating autophagy and inhibiting tumor formation and proliferation [67]. Autophagy is involved in oncogenesis as it affects the adaptation of tumoral cells to stress conditions, such as ischemia, where it is localized in the tumor center to support malignant cells with the essential nutrients for their expansion prior to the initiation of angiogenesis [68]. Moreover, the loss of the tumor repressor enzyme phosphatase and tensin analog (PTEN) activates Akt, which results in a significant reduction in the autophagy of malignant cells [69].
Notably, in MM, autophagy might serve as a pro-survival strategy that helps tumor cells to remove the enormous accumulation of harmful, misfolded Igs. Additionally, autophagy helps myeloma cells resist proteasome antagonists. Proteasome inhibition causes ER overloading and stress by increasing the aggregation of damaged proteins in the intracellular milieu. Proteasome inhibition also promotes autophagic activity, resulting in drug resistance, which is consistent with the close relationship between cell stress, autophagy, and apoptosis. Hence, a novel treatment approach that targets autophagy to trigger the death of myeloma cells and enhance drug susceptibility may be considered. Particularly, targeting autophagy may concentrate on activating or inhibiting autophagic activity due to its dual function as a process of pro-survival or cell death [70]. Moreover, various stress factors in the tumor microenvironment, such as hypoxia, starvation, inflammation, and extracellular matrix breakdown, activate pro-survival autophagy.
The evolution of chemoresistance in malignancies is a significant issue in the clinic. The development of autophagy inhibitors, intended to boost chemosensitivity, has been hugely influenced by this [71]. Phase I/II clinical trials using hydroxychloroquine (HCQ)-mediated autophagy repression have been performed recently in a number of malignancies, such as myeloma, pancreatic cancer, and melanoma [72][73][74][75]. These studies assessed patient toxicity, clinical activity, maximum tolerated dosages, and pharmacodynamics. It has been demonstrated that the antimalarial medicine HCQ inhibits the end stage of autophagic activity [76]. In a phase I clinical trial, the mTOR inhibitor temsirolimus and HCQ were used to treat patients with melanoma and advanced solid tumors. Although there was no response, the majority of patients who received treatment had stable illnesses. The median progression-free survival in 13 patients with melanoma who received HCQ at 1200 mg/d in addition to temsirolimus was 3.5 months [73]. Moreover, pancreatic adenocarcinoma was the subject of HCQ clinical studies during which HCQ had a minor anti-tumor effect due to the unstable inhibition of autophagy [74]. Due to its efficiency, the proteasome inhibitor bortezomib has become the standard of care treatment for multiple myeloma [77]. In 22 individuals with refractory or relapsed myeloma, bortezomib and HCQ produced 14% partial responses, 14% modest responses, and 45% persistent illness [75]. Consistent with other studies, the HCQ with bortezomib combination led to an elevation of autophagosomes as a pharmacodynamic indicator of autophagic activity modification. The abovementioned clinical trials demonstrate that it is possible to achieve therapy-induced autophagy suppression and that significant advancements have been made in the manipulation of autophagy for cancer treatment.
4. Multiple Myeloma Drug Resistance and Autophagy
Various stress conditions, including hypoxia, starvation, extracellular matrix reduction, and inflammation in the tumor tissue, activate survival-promoting autophagy. The emergence of proteasome inhibitors (PIs) and immunomodulatory drugs has significantly developed the prognosis of MM patients. Bortezomib prevents MM cell proliferation, leads to apoptosis, and disturbs MM cell crosstalk with the BM stroma by inhibiting cytokine circuits [78][79]. Even though the efficacy of bortezomib is proven in MM patients, relapse caused by bortezomib resistance is unavoidable, and the malignancy is still untreatable [80]. Bortezomib resistance is thought to be caused by the induction of autophagy, as bortezomib increases the aggregation of polyubiquitinated proteins [63][80]. Protein aggregation leads to the formation of aggresomes and autophagosomes, which may increase protein degradation, tumor viability, and relative resistance to drugs (Figure 3). Bortezomib interacts with cancer-associated fibroblasts (CAFs) in the BM microenvironment to trigger ROS and autophagy by blocking mTOR and p62 [63][80]. CAFs are critical in the BM microenvironment and enhance cancer formation, development, and drug resistance. In vitro co-cultures of MM CAFs and MM cells resist bortezomib, indicating that MM CAFs inhibit the apoptosis caused by bortezomib [81].
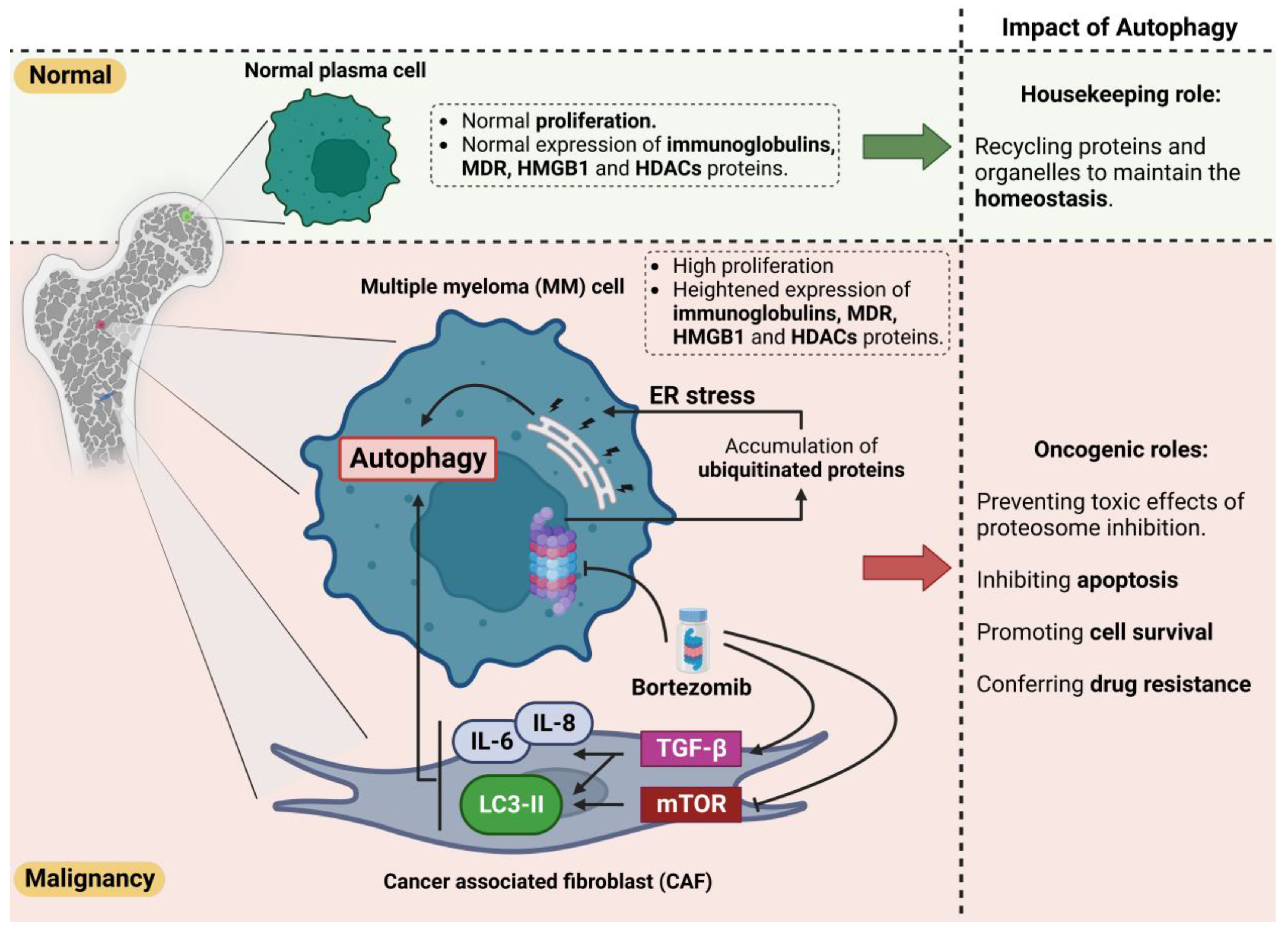
Figure 3. Dual role of autophagy in normal and malignancy conditions. Autophagy plays a protective role in the homeostasis of normal cells via recycling of the aggregated/unfolded/misfolded proteins and defective organelles. However, this protective effect could enhance the survival of tumor cells by clearing the accumulated proteins and improving their turnover, thereby inhibiting apoptosis. Moreover, autophagy could cause resistance to bortezomib by inducing ER stress in MM cells and triggering the pro-autophagic pathways in cancer-associated fibroblast cells (CAFs). Heightened autophagy clears the bortezomib-induced accumulation of ubiquitinated proteins. The MM cells could subsequently escape from apoptotic pathways and survive during treatment.
Moreover, bortezomib treatment has been shown to promote autophagy in myeloma CAFs by inhibiting p62 and mTOR, inducing LC3, and activating TGF-b [63] (Figure 3). Enhanced autophagy prevents the harmful effects of proteasome inhibition and inhibits apoptosis. This potential drug resistance is an essential obstacle to therapy and the survival of individuals with MM. As discussed earlier, no treatment for MM is known so far. As resistance to bortezomib evolves, scientists are seeking approaches to conquer this problem by understanding major resistance strategies as potential therapeutic targets [82]. Regarding the pathophysiology of resistance to bortezomib, autophagy is crucial in preventing apoptosis, despite enhanced stress [80]. As mentioned earlier, caspase 10 is an autophagy regulator and inhibits cell death induced by autophagy [83]. Once the mechanism for attenuating the pro-survival effects of autophagy is compromised, the autophagy triggered by bortezomib treatment and resistance might result in cell death.
Since bortezomib was approved in 2003 to be used in the therapies of resistant/relapsed multiple myeloma (MM) [84], and with the subsequent agreement to apply bortezomib as the first-line treatment of MM in 2008, the use of PIs for therapeutic purposes in blood cancers has increased dramatically [85]. In comparison with standard chemotherapy strategies, patients with MM malignancy lived twice as long after the development of autologous stem cell transplantation [86]. Bortezomib was authorized in 2006 to treat individuals with relapsed mantle cell lymphoma (MCL) [87]. Later, new PIs were produced to improve bortezomib’s oral bioavailability, lessen unfavorable effects, and address resistance to bortezomib occurrence [88]. Then, the new PI carfilzomib was subsequently authorized to treat MM individuals who relapsed after receiving bortezomib plus an immunomodulatory medication (IMiD) [89][90]. Encouraging outcomes of PIs in individuals with MM and MCL pave the way for testing their action in more hard-to-treat hematologic cancers having low survivability, such as acute leukemia [91]. In spite of the positive outcomes of bortezomib therapy, the emergence of the bortezomib resistance phenomenon is a growing barrier that hampers the drug’s treatment efficacy [92][93][94]. Therefore, understanding and managing the complex processes behind both innate and adaptive resistance to PIs is essential to improve their therapeutic effectiveness. Alongside proteasomal activity, autophagy provides an alternate approach to recycling and breaking down proteins inside the cells. Autophagy and ubiquitin–proteasome system (UPS) were formerly believed to act separately, but it has recently been discovered that these two proteolytic mechanisms work together. In order to degrade cytosolic proteins, autophagy acts through the fusion of lysosomes with double-layer structures called autophagosomes. Numerous human diseases, including cancer and neurological disorders, are related to disturbed autophagy [95]. It seems that autophagy functions as a tumor suppressor in the normal state, while it can promote tumor cell viability during stress situations [96]. It appears that by inhibiting the proteasome, autophagy is activated as a survival strategy for the removal of UPS substrates [97]; therefore, activated autophagy may be involved in the resistance to bortezomib [98]. Accordingly, activating transcription factor 4 (ATF4), which induces autophagic activity, was elevated after the proteasome was inhibited [99][100][101].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24076019
References
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721.
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741.
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534.
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873.
- Kunz, J.B.; Schwarz, H.; Mayer, A. Determination of four sequential stages during microautophagy in vitro. J. Biol. Chem. 2004, 279, 9987–9996.
- Jardon, M.A.; Rothe, K.; Bortnik, S.; Vezenkov, L.; Jiang, X.; Young, R.N.; Lum, J.J.; Gorski, S.M. Autophagy: From Structure to Metabolism to Therapeutic Regulation; Taylor & Francis: Abingdon, UK, 2013.
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417.
- Arias, E. Methods to study chaperone-mediated autophagy. Methods Enzymol. 2017, 588, 283–305.
- Schneider, J.L.; Cuervo, A.M. Autophagy and human disease: Emerging themes. Curr. Opin. Genet. Dev. 2014, 26, 16–23.
- Cuervo, A.M.; Dice, J.F.; Knecht, E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem. 1997, 272, 5606–5615.
- Nikesitch, N.; Rebeiro, P.; Ho, L.L.; Pothula, S.; Wang, X.M.; Khong, T.; Quek, H.; Spencer, A.; Lee, C.S.; Roberts, T.L. The role of chaperone-mediated autophagy in Bortezomib resistant multiple myeloma. Cells 2021, 10, 3464.
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473.
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41.
- Klionsky, D.J. The molecular machinery of autophagy: Unanswered questions. J. Cell Sci. 2005, 118, 7.
- Vakifahmetoglu-Norberg, H.; Xia, H.-G.; Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Investig. 2015, 125, 5–13.
- Kenific, C.M.; Debnath, J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015, 25, 37–45.
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132.
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295.
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584.
- Rowinsky, E.K. Targeting the molecular target of rapamycin (mTOR). Curr. Opin. Oncol. 2004, 16, 564–575.
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603.
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293.
- Díaz-Troya, S.; Pérez-Pérez, M.E.; Florencio, F.J.; Crespo, J.L. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 2008, 4, 851–865.
- Kwon, G.; Marshall, C.A.; Pappan, K.L.; Remedi, M.S.; McDaniel, M.L. Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes 2004, 53, S225–S232.
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484.
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003.
- Lozy, F.; Karantza, V. Autophagy and cancer cell metabolism. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2012; pp. 395–401.
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596.
- Simonsen, A.; Tooze, S.A. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 2009, 186, 773–782.
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007, 282, 13123–13132.
- Kang, R.; Zeh, H.; Lotze, M.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580.
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12.
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124.
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145.
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880.
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023.
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80.
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56.
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720.
- Rosenfeldt, M.T.; Ryan, K.M. The role of autophagy in tumour development and cancer therapy. Expert Rev. Mol. Med. 2009, 11, e36.
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 1516–1523.
- Dong, Z.; Liang, S.; Hu, J.; Jin, W.; Zhan, Q.; Zhao, K. Autophagy as a target for hematological malignancy therapy. Blood Rev. 2016, 30, 369–380.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30.
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D. Global burden of multiple myeloma: A systematic analysis for the global burden of disease study 2016. JAMA Oncol. 2018, 4, 1221–1227.
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417.
- Weiss, B.M.; Abadie, J.; Verma, P.; Howard, R.S.; Kuehl, W.M. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 2009, 113, 5418–5422.
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249.
- Rajkumar, S.V.; Landgren, O.; Mateos, M.V. Smoldering multiple myeloma. Blood 2015, 125, 3069–3075.
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346.
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046.
- Jelinek, T.; Kryukov, F.; Rihova, L.; Hajek, R. Plasma cell leukemia: From biology to treatment. Eur. J. Haematol. 2015, 95, 16–26.
- Ravi, P.; Kumar, S.K.; Roeker, L.; Gonsalves, W.; Buadi, F.; Lacy, M.Q.; Go, R.S.; Dispenzieri, A.; Kapoor, P.; Lust, J.A.; et al. Revised diagnostic criteria for plasma cell leukemia: Results of a Mayo Clinic study with comparison of outcomes to multiple myeloma. Blood Cancer J. 2018, 8, 116.
- Milan, E.; Fabbri, M.; Cenci, S. Autophagy in plasma cell ontogeny and malignancy. J. Clin. Immunol. 2016, 36, 18–24.
- Hoang, B.; Benavides, A.; Shi, Y.; Frost, P.; Lichtenstein, A. Effect of autophagy on multiple myeloma cell viabilityAutophagy and Myeloma. Mol. Cancer Ther. 2009, 8, 1974–1984.
- Caro, L.H.P.; Plomp, P.J.; Wolvetang, E.J.; Kerkhof, C.; Meijer, A.J. 3-Methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. Eur. J. Biochem. 1988, 175, 325–329.
- Wang, G.; Zhou, P.; Chen, X.; Zhao, L.; Tan, J.; Yang, Y.; Fang, Y.; Zhou, J. The novel autophagy inhibitor elaiophylin exerts antitumor activity against multiple myeloma with mutant TP53 in part through endoplasmic reticulum stress-induced apoptosis. Cancer Biol. Ther. 2017, 18, 584–595.
- Lamy, L.; Ngo, V.N.; Emre, N.T.; Shaffer, A.L.; Yang, Y.; Tian, E.; Nair, V.; Kruhlak, M.J.; Zingone, A.; Landgren, O. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 2013, 23, 435–449.
- Yun, Z.; Zhichao, J.; Hao, Y.; Ou, J.; Ran, Y.; Wen, D.; Qun, S. Targeting autophagy in multiple myeloma. Leuk. Res. 2017, 59, 97–104.
- Jaganathan, S.; Malek, E.; Vallabhapurapu, S.; Vallabhapurapu, S.; Driscoll, J.J. Bortezomib induces AMPK-dependent autophagosome formation uncoupled from apoptosis in drug resistant cells. Oncotarget 2014, 5, 12358.
- Riz, I.; Hawley, T.S.; Hawley, R.G. KLF4-SQSTM1/p62-associated prosurvival autophagy contributes to carfilzomib resistance in multiple myeloma models. Oncotarget 2015, 6, 14814.
- Hoang, B.; Benavides, A.; Shi, Y.; Frost, P.; Lichtenstein, A. Abstract# 1877: Impact of autophagy on multiple myeloma cell viability. Cancer Res. 2009, 69, 1877.
- Fu, Y.-F.; Liu, X.; Gao, M.; Zhang, Y.-N.; Liu, J. Endoplasmic reticulum stress induces autophagy and apoptosis while inhibiting proliferation and drug resistance in multiple myeloma through the PI3K/Akt/mTOR signaling pathway. Oncotarget 2017, 8, 61093.
- Frassanito, M.; De Veirman, K.; Desantis, V.; Di Marzo, L.; Vergara, D.; Ruggieri, S.; Annese, T.; Nico, B.; Menu, E.; Catacchio, I. Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia 2016, 30, 640–648.
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.-T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood J. Am. Soc. Hematol. 2012, 120, 3519–3529.
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Hassan, H.; Zhao, J.; Carew, J.S.; Nawrocki, S.T.; et al. Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia-Pac. J. Clin. Oncol. 2021, 17, 193–208.
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676.
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.-H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–698.
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64.
- Aquila, S.; Santoro, M.; Caputo, A.; Panno, M.L.; Pezzi, V.; De Amicis, F. The Tumor Suppressor PTEN as Molecular Switch Node Regulating Cell Metabolism and Autophagy: Implications in Immune System and Tumor Microenvironment. Cells 2020, 9, 1725.
- Desantis, V.; Saltarella, I.; Lamanuzzi, A.; Mariggiò, M.; Racanelli, V.; Vacca, A.; Frassanito, M. Autophagy: A new mechanism of prosurvival and drug resistance in multiple myeloma. Transl. Oncol. 2018, 11, 1350–1357.
- Ndoye, A.; Weeraratna, A.T. Autophagy—An emerging target for melanoma therapy. F1000Research 2016, 5, 1888.
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379.
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402.
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638.
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390.
- Solomon, V.R.; Lee, H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009, 625, 220–233.
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: Further developments. J. Hematol. Oncol. 2020, 13, 159.
- Hamedi, K.R.; Harmon, K.A.; Goodwin, R.L.; Arce, S. Autophagy and the Bone Marrow Microenvironment: A Review of Protective Factors in the Development and Maintenance of Multiple Myeloma. Front. Immunol. 2022, 13, 889954.
- RI, M. Mechanism of action of bortezomib in multiple myeloma therapy. Int. J. Myeloma 2016, 6, 1–6.
- Yan, Y.; Chen, X.; Wang, X.; Zhao, Z.; Hu, W.; Zeng, S.; Wei, J.; Yang, X.; Qian, L.; Zhou, S. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 171.
- De Veirman, K.; Rao, L.; De Bruyne, E.; Menu, E.; Van Valckenborgh, E.; Van Riet, I.; Frassanito, M.A.; Di Marzo, L.; Vacca, A.; Vanderkerken, K. Cancer associated fibroblasts and tumor growth: Focus on multiple myeloma. Cancers 2014, 6, 1363–1381.
- Murray, M.Y.; Zaitseva, L.; Auger, M.J.; Craig, J.I.; MacEwan, D.J.; Rushworth, S.A.; Bowles, K.M. Ibrutinib inhibits BTK-driven NF-κB p65 activity to overcome bortezomib-resistance in multiple myeloma. Cell Cycle 2015, 14, 2367–2375.
- Martin, S. Autophagy in Multiple Myeloma: What Makes You Stronger Can Also Kill You. Cancer Cell 2013, 23, 425–426.
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade®: US FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513.
- McBride, A.; Ryan, P.Y. Proteasome inhibitors in the treatment of multiple myeloma. Expert Rev. Anticancer Ther. 2013, 13, 339–358.
- San Miguel, J.F.; Mateos, M.V. Can multiple myeloma become a curable disease? Haematologica 2011, 96, 1246–1248.
- Kane, R.C.; Dagher, R.; Farrell, A.; Ko, C.-W.; Sridhara, R.; Justice, R.; Pazdur, R. Bortezomib for the treatment of mantle cell lymphoma. Clin. Cancer Res. 2007, 13, 5291–5294.
- Kirk, C.J. Discovery and development of second-generation proteasome inhibitors. In Seminars in Hematology; Elsevier: Amsterdam, The Netherlands, 2012; pp. 207–214.
- Dimopoulos, M.A.; San-Miguel, J.F.; Anderson, K.C. Emerging therapies for the treatment of relapsed or refractory multiple myeloma. Eur. J. Haematol. 2011, 86, 1–15.
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.D.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J. US Food and Drug Administration Approval: Carfilzomib for the Treatment of Multiple MyelomaFDA Approval Summary of Carfilzomib. Clin. Cancer Res. 2013, 19, 4559–4563.
- Niewerth, D.; Dingjan, I.; Cloos, J.; Jansen, G.; Kaspers, G. Proteasome inhibitors in acute leukemia. Expert Rev. Anticancer Ther. 2013, 13, 327–337.
- Ahn, J.-S.; Jung, S.-H.; Yang, D.-H.; Bae, S.-Y.; Kim, Y.-K.; Kim, H.-J.; Lee, J.-J. Patterns of relapse or progression after bortezomib-based salvage therapy in patients with relapsed/refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2014, 14, 389–394.
- Cheriyath, V.; Jacobs, B.S.; Hussein, M.A. Proteasome inhibitors in the clinical setting: Benefits and strategies to overcome multiple myeloma resistance to proteasome inhibitors. Drugs R D 2007, 8, 1–12.
- Petrucci, M.T.; Giraldo, P.; Corradini, P.; Teixeira, A.; Dimopoulos, M.A.; Blau, I.W.; Drach, J.; Angermund, R.; Allietta, N.; Broer, E. A prospective, international phase 2 study of bortezomib retreatment in patients with relapsed multiple myeloma. Br. J. Haematol. 2013, 160, 649–659.
- Lilienbaum, A. Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 2013, 4, 1.
- White, E.; DiPaola, R.S. The Double-Edged Sword of Autophagy Modulation in CancerAutophagy in Cancer Therapy. Clin. Cancer Res. 2009, 15, 5308–5316.
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524.
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.-M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666.
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A.L. The Role of ATF4 Stabilization and Autophagy in Resistance of Breast Cancer Cells Treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423.
- Zang, Y.; Thomas, S.M.; Chan, E.T.; Kirk, C.J.; Freilino, M.L.; DeLancey, H.M.; Grandis, J.R.; Li, C.; Johnson, D.E. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin. Cancer Res. 2012, 18, 5639–5649.
- Zhu, K.; Dunner, K.; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462.
This entry is offline, you can click here to edit this entry!