Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Electrochemistry
A stable life support system in the spacecraft can greatly promote long-duration, far-distance, and multicrew manned space flight. Therefore, controlling the concentration of CO2 in the spacecraft is the main task in the regeneration system. The electrocatalytic CO2 reduction can effectively treat the CO2 generated by human metabolism. This technology has potential application value and good development prospect in the utilization of CO2 in the space station.
- carbon dioxide
- electrocatalytic reduction
- electrocatalyst
- electrolyte
- reactor design
- spacecraft life support system
1. Introduction
With the continuous development of human science and technology, space exploration will face unprecedented opportunities and challenges [1,2,3,4,5]. Currently, realizing the recycle of materials in closed space stations is the best solution to solve the problem of water (H2O) and oxygen (O2) resupply faced by human activities, which not only can extend the duration of manned missions and reduce transportation costs, but also is a prerequisite for interstellar migration [6,7,8,9,10]. As the end product of human respiration and metabolic processes, carbon dioxide (CO2) carries a large amount of oxygen [11,12,13]. Therefore, recovering O2 from waste CO2 is an important way to achieve resource recycling and reduce transportation costs for medium- and long-term manned extraterrestrial missions [14,15,16,17,18].
The concentration of CO2 in the space station directly affects the life and health system of the astronauts [19]. To reduce the concentration of CO2 (<0.5%) and maintain the balance of air components, it is necessary to treat the CO2 emissions at 0.7–1.0 kg per person per day. How to better transform and recycle CO2 in the space station has become the focus of researchers [20,21,22,23]. The methods of CO2 transformation can be classified into two categories: physical absorption and chemical conversion [24,25]. The chemical conversion mainly includes thermochemistry, photocatalytic reduction, electrocatalytic reduction, and photoelectrocatalytic reduction [26,27,28,29]. Chemical conversion can capture and immobilize CO2 or convert it into useful low-carbon fuels, such as CO, CH4, HCOOH, CH3OH, etc. [30,31,32,33,34].
2. The Mechanism of Electrocatalytic Reduction of CO2
The electrocatalytic reduction of CO2 often occurs at the interface between the electrode and the electrolyte, and the electrolyte is usually an aqueous solution of potassium bicarbonate [19]. As shown in Figure 1, the multiphase catalytic process generally consists of four main steps: (i) The CO2 in the solution diffuses to the surface of the working electrode. (ii) The electrocatalyst adsorbs CO2 from the solution. (iii) Electron transfer or proton migration is used to cleave C-O bonds or form C-H bonds. (iv) The generated products are detached from the electrocatalyst surface and diffused into the electrolyte [60,61]. The adsorption of CO2 on the electrocatalyst surface is an important step in the whole reaction. During the adsorption process, the C=O bonding is strongly perturbed by the substrate, and the electrons are shared between the CO2 and the catalyst. A number of reaction mechanisms have been proposed for the conversion of CO2. It is widely accepted that the reactive species are the neutral hydrated CO2 molecules, and they will convert into CO2•− in the adsorption process on most of the main-group metal electrodes in aqueous media. Then the absorbed CO2•− reacts with the H2O molecules to form HCO2•. The intermediate would convert into to HCO2− because of its unstable unpaired electron, followed by the desorption of HCO2− species [60].
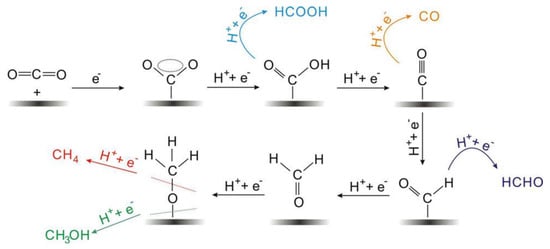
Figure 1. Reaction mechanism of electrochemical CO2 reduction on electrodes in aqueous solutions and the formation paths for the five main C1 products during the reduction process.
3. Electrocatalysts
Over the past few decades, researchers have attempted to develop high-performance CO2 electrocatalysts and have carried out extensive work [71,72,73]. The catalysts could reduce the activation energy required for the electroreduction of CO2, thereby minishing the reduction overpotential and current density. Most of the studies have focused on transition-metal-based catalysts, which are mainly Cu, Au, Fe, Ag, Re, Mn, Co, Ni, Pd, Ir, Ru, etc. [17,74,75,76]. The main forms of the complexes include transition metal polypyridines, metal porphyrins/phthalocyanines, and various metal phosphine complexes [77,78].
Among the transition metal complexes, Cu complexes had better activity. Recently, Raja et al. [78] designed a dinuclear Cu (I) complex as catalysts for the electroreduction of CO2, and its catalytic cycle is shown in Figure 2 below. They found that the Cu (I) system could be oxidized by CO2, which suggested that the selective bonding of CO2 and Cu (I) ions could provide a low-energy pathway for the formation of CO2•− radical anions. Thus, the Cu (II) tetranuclear oxalate-bridged complex [2]4+ was thermodynamically favorable. Moreover, the bonding of CO2 to the Cu(I) center in the dinuclear copper (I) complex [1]2+ was faster and has a higher selectivity. This was due to the low solubility of lithium oxalate in acetonitrile, and the release of the oxalate double anion from [2]4+ in the presence of LiClO4 could be accomplished instantaneously, and the dinuclear copper (II) complex [4]4+ was produced. Consequently, the electrocatalytic reduction of Cu (II) ions to Cu (I) might be the rate-controlling step in this system. The electrode surface was coated by the lithium oxalate generated during the reaction and then prevented the effective electron transfer.
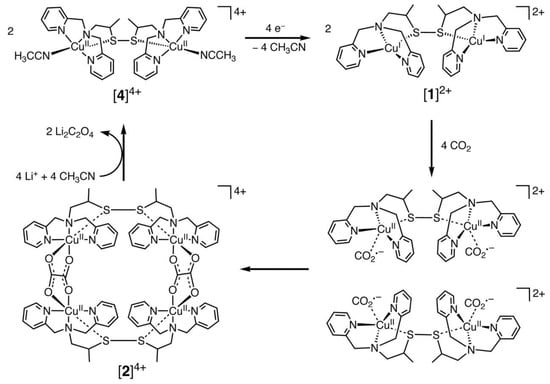
Figure 2. Proposed electrocatalytic cycle for oxalate formation.
Cu electrodes have good catalytic performance for the conversion of CO2 to alkanes and alcohols [64]. The moderate hydrogen evolution overpotential of Cu electrodes can properly suppress the hydrogen production. Therefore, Cu electrodes can generate relatively high current efficiency [108]. Recently, Matthew et al. [109] prepared Cu-modified electrodes by calcining Cu sheets in air and then further electrochemically reducing and calcining the generated Cu2O (Figure 3). The activity exhibited by such electrodes in reducing CO2 was highly dependent on the initial thickness of the Cu2O layer. The experimental results showed that the activity of the electrodes prepared from a Cu2O thin layer at 130 °C was not different from that of polycrystalline Cu. However, the Cu2O electrode formed by calcination at 500 °C with a thickness of not less than 3 μm had a large roughness coefficient, and its overpotential was 0.5 V smaller than that of polycrystalline Cu in the reduction of CO2. More significantly, the current density of this electrode would be higher than 1 mA/cm2 at an overpotential lower than 0.4 V, which was larger than the reported activity of other metal electrodes. Meanwhile, the Cu-modified electrode obtained by calcination at 500 °C remained stable after reacting for 7 h, while the polycrystalline Cu electrode started to passivate within 1 h under the same circumstances.
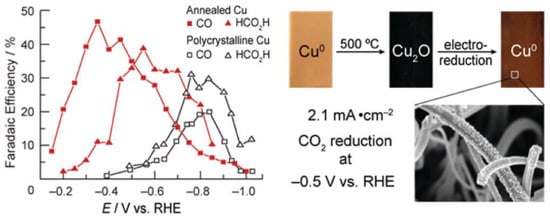
Figure 3. Electrocatalytic performance and the preparation process of Cu electrodes.
To confirm that different components of binary nanocrystals have different effects on the performance of the electrocatalytic reduction of CO2, Guo et al. [110] investigated the performance of Cu-Pt binary alloy nanocrystals for the electrochemical reduction of CO2 in 0.5 M KHCO3 at room temperature. As shown in Figure 4, it was found that among Cu-Pt nanocrystals with different molar ratios, the catalyst with a molar ratio of 3:1 had the best activity for the catalytic reduction of CO2, the lowest onset potential (−0.972 V), and the highest current density (0.598 mA/cm−2, −1.3 V vs. SCE). The optimization of catalyst components was compared, and a reasonable mechanism hypothesis was proposed. As illustrated in Figure 5, Pt had a unique adsorption capacity for protons, and it was an efficient catalyst for HER, leading that H2O molecules in solution would be adsorbed on Pt atoms in Cu-Pt nanocrystals. CO*, the reaction intermediate generated by CO2 gaining electrons, was adsorbed on Cu atoms, and then combined with H+ provided by H2O to generate HCO* and finally CH4. The above mechanism well explains why Cu-Pt nanocrystals with a molar ratio of 3:1 had the highest activity in the catalytic reduction of CO2. In the process of CH4 generation, CO* and H+ were indispensable, and Cu promoted CO2 to gain electrons to generate CO*. Pt facilitated the adsorption of H2O to generate H+, so the molar ratio of Cu-Pt had an optimal value; too much Pt or Cu was not conducive to CH4 production.
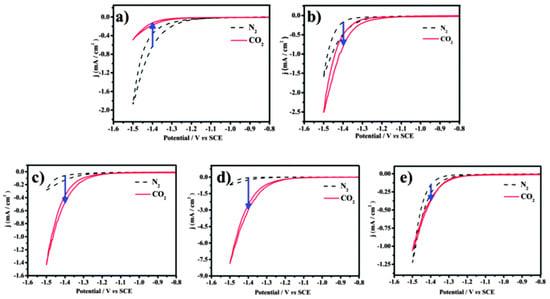
Figure 4. The cyclic voltammograms of catalysts were recorded in N2- and CO2-saturated 0.5 M KHCO3 with a scan rate of 10 mV/s−1 between −0.8 and −1.5 V (vs. SCE). (a) Cu-Pt-1#, (b) Cu-Pt-2#, (c) Cu-Pt-3#, (d) Cu-Pt-4#, and (e) Cu-Pt-5#.
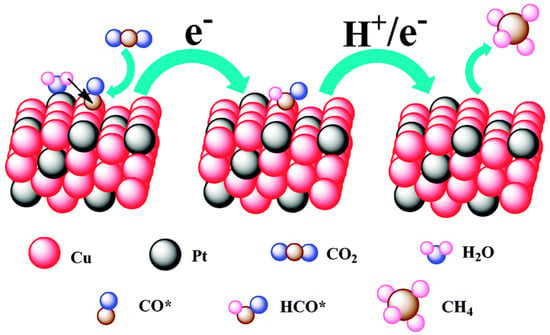
Figure 5. A proposed mechanism illustrating the steps of CO2 electroreduction and CH4 formation occurring at the Cu-Pt 3:1 NC catalyst.
However, Cu-based catalysts still faced some problems because the instability of Cu materials was easily deactivated and decomposed during the catalytic reduction of CO2. Some prepared Cu-based catalysts were easily oxidized when exposed to air [21,75,109]. The valence and morphology changes of Cu-based catalysts during the catalytic process should be deeply elucidated, which conduced to provide a crucial role. Ni et al. [111] proposed a modulation strategy: hydrogen reduction valence, which could simply regulate the ratio of different valence states of Cu by optimizing the reduction time, and helped to investigate the mechanism of multivalent Cu in depth. The experimental results showed that the excellent performance of G-CuxO-2h not only stabilized the intermediate product CO2•−, but also accelerated the rate-limiting step in the HCOO− desorption process, which was related to the optimal Cu (I) content in the catalyst. The paper also proposed a “buffering effect” to explain the stability of G-CuxO-2h, as shown in Figure 6. Cu (II) from the thicker subsurface layer acted as a sacrificial source to supplement Cu (I), thus balancing the Cu (I) content in the surface layer and maintaining the activity of the catalyst in the reaction.
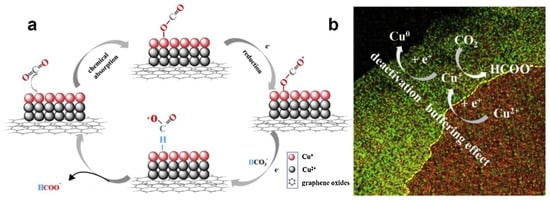
Figure 6. (a) A proposed mechanism illustrating the steps of electrochemical CO2 reduction and the formation of HCOOH occurring at the G-CuxO-2 h electrocatalyst. (b) A proposed “buffering effect” illustrating the origin of the durability of G-CuxO-T electrocatalysts.
Furthermore, the electrocatalytic reduction of CO2 at the Au electrode has also been investigated by Matthew et al. [112]. They used the periodically symmetric square wave potential method to prepare an amorphous Au2O3 layer on a Au electrode. This electrode was used directly for the electrocatalytic reduction of CO2, in which Au2O3 was reduced to Au within 15 min. The electrode exhibited high selectivity for the product CO in the reduction of CO2 with an overpotential of only 0.14 V, and the activity was maintained for at least 8 h. The authors attributed the high activity of such catalysts to the increased stability of the CO2•− intermediate; the electrolyte HCO3− acted as H+ donors during the catalytic process (Figure 7).
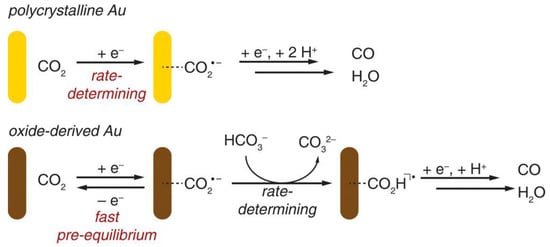
Figure 7. Proposed mechanisms for CO2 reduction to CO on polycrystalline Au and oxide-derived Au.
Gao et al. synthesized a Pd nanoparticle electrode and efficiently catalyzed the reduction of CO2 to CO. They found that Pd nanoparticle size exhibited significant size dependence in the range of 2.4–10.3 nm [113]. With the Pb nanoparticle size changes from 10.3 to 3.7 nm, the Faraday conversion efficiency of CO generation at −0.89 V increased from 5.8% to 91.2%, while the current density of CO generation was enhanced 18.4 times (Figure 8). The relationship between catalyst performance and particle size was obtained using the density functional theory (DFT) to further analyze the free energy of CO2 reduction and HER at three different reaction sites: plane, step, and corner. The results indicated that the relationship between the conversion frequency (TOF) of CO generation and catalyst particle size presented a volcano-like curve (Figure 9). This illustrated that CO2 adsorption, COOH* formation, and CO* removal could be modulated by changing the size of Pb nanoparticles, thus enabling the transition of Pd nanoparticles from HER catalysts to efficient CO2 reduction catalysts.
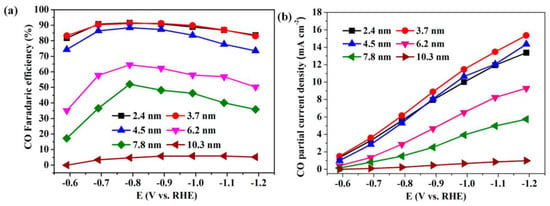
Figure 8. Applied potential dependence of (a) Faradaic efficiencies and (b) current densities for CO production over Pd NPs with different sizes.
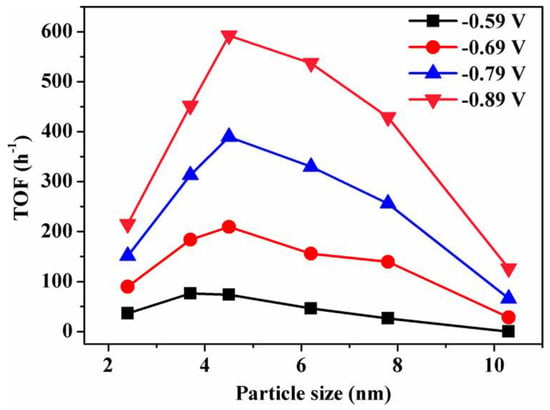
Figure 9. Size dependence of TOF for CO production on Pd NPs at various potentials.
As well known, the metal electrode possessed high activity for the catalytic reduction of CO2 [114,115]. The outstanding catalytic activity was obtained by reducing metal oxides to metal electrode [116,117,118]. This electrode could decrease the reduction potential of CO2 to a thermodynamic minimum. In comparison, other preparation methods caused many microstructural changes in the catalysts, such as interfaces and defects [119,120,121]. The influence of metal oxides on the catalytic performance of metals was related to these microstructures. However, the mechanism of the above system was not clear [122,123]. Recently, a 4-atomic-layer-thick Co oxidized heterostructure (see Figure 10) was prepared by Gao et al. by means of ligand-confined growth and used to explain the effect of metal surface oxides on their own electroreduction CO2 properties [124]. This work demonstrated that metal atoms located in specific oxidation states enabled the production of higher catalytic activity through specific arrangements, thus providing a new idea for the development of efficient and stable catalysts for CO2 reduction. It was important for promoting research on the mechanism of metal oxides’ electrocatalytic reduction of CO2.
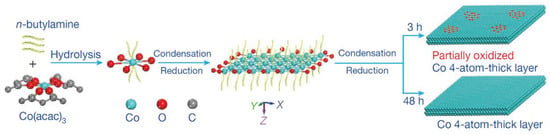
Figure 10. Schematic formation process of the partially oxidized and pure-Co 4-atomic-layer, respectively.
Transition metal porphyrins have also shown superior reactivity in catalysts for the electrocatalytic reduction of CO2, such as iron porphyrins and cobalt porphyrins [125]. Cyrille et al. found that the modified iron porphyrins accelerated the reduction reaction of CO2 after the introduction of phenolic groups in all the neighbors of the iron porphyrin phenyl group. It was concluded that the increased activity of iron porphyrins was related to the high local concentration of protons associated with the phenolic hydroxyl substituents; the relationship between the high turnover frequency (TOF) and the small overpotential (η) of the catalyst was systematically investigated (Figure 11) [77].
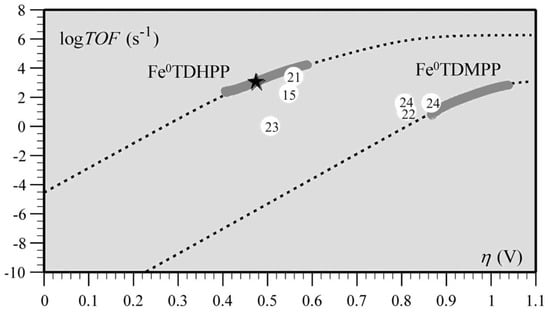
Figure 11. Correlation between turnover frequency and overpotential for the series of CO2-to-CO electroreduction catalysts. The star indicates TOF and η values from preparative-scale experiments of Fe0TDHPP.
Cobalt porphyrins as catalysts for the electrocatalytic reduction of CO2 have a relatively high overpotential (−1.3~−1.6 V vs. SHE) and a low TOF. The possible catalytic mechanism of cobalt porphyrins was analyzed by Kevin et al. using DFT calculations [126]. From the calculations, it was clear that CO2 was bonded to the particle [Co(I)P]−, and the mechanism is shown in Figure 12. Besides, the presence of water played a crucial role because the key intermediates [Co(I)P-CO2]2− and [Co(II)P-CO2H]− were stabilized by hydrogen bonding interaction, which caused the exothermic breakage of the C-O bond. Theoretical calculations also revealed that the electron transfer between the gas diffusion electrode and the polymerized porphyrin catalyst was the rate-controlling step of the whole reaction. These findings were important implications for the capture and the electrochemical reduction of CO2, respectively.
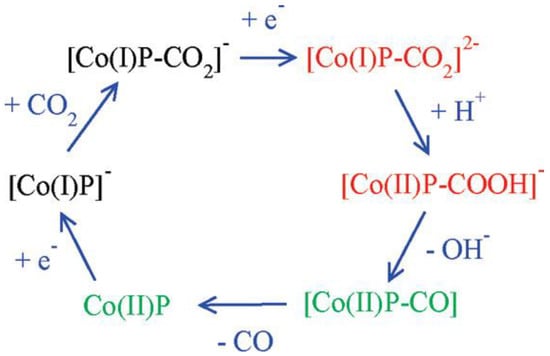
Figure 12. Mechanism of CO2 reduction with electron addition deduced from hybrid DFT plus dielectric continuum redox potential calculations. Red denotes key intermediates; green species should undergo fast reactions.
The effect of organometallic Ag catalysts supported by carbon–nitrogen in the electrochemical conversion of CO2 was investigated by Claire et al. [127]. It was shown that such catalysts decreased the reaction overpotential and improved the selectivity, which also led to an enhanced reaction rate for CO2 reduction. Moreover, these nitrogen-containing compounds might act as cocatalysts in the electrochemical reduction of CO2, create a coordinated effect with the Ag surface, and thus facilitate electron transfer. Remarkably, they found that not all nitrogen-containing atoms introduced to the carbon surface exhibited the properties of such catalysts. Therefore, to further enhance the activity and selectivity, a more in-depth study of the CO2 reduction mechanism was needed to elucidate the interactions between these complexes and the carbon substrate.
Agarwal’s group carried out an in-depth study of the CO2 reduction mechanism. Although the results were based on the photocatalytic reduction of CO2, their systematic analysis of the CO2 reduction mechanism was equally applicable in the direction of electrocatalytic reduction of CO2 [128]. First, they found that tricarbonyl Re complexes, such as Re(bpy)(CO)3Cl (bpy=2,2′-dipyridine), exhibited high activity in catalytic CO2 reduction in the presence of electron sacrificial agents. Subsequently, they investigated the potential pathway of formate generation by the Re-hydride insertion theory in the presence of triethylamine (TEA) using DFT. It was suggested that TEA was the main donor of hydrogen atoms and electrons, and its catalytic cycle pathway is shown in Figure 13.
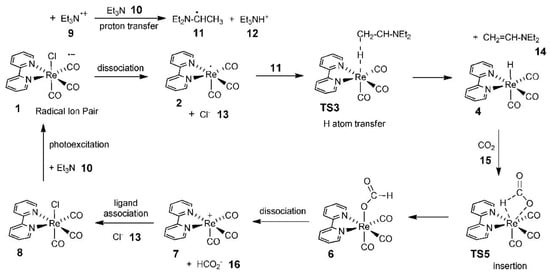
Figure 13. Computed photocatalytic cycle for CO2 reduction on a Re catalyst.
This entry is adapted from the peer-reviewed paper 10.3390/catal13040644
This entry is offline, you can click here to edit this entry!