Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Rheumatology
Systemic lupus erythematosus (SLE) is a genetically predisposed, female-predominant disease, characterized by multiple organ damage, that in its most severe forms can be life-threatening. The pathogenesis of SLE is complex and involves cells of both innate and adaptive immunity. The distinguishing feature of SLE is the production of autoantibodies, with the formation of immune complexes that precipitate at the vascular level, causing organ damage.
- systemic lupus erythematosus
- B-cells
- T-cells
- plasmacytoid cells
1. The Role of Adaptive Immunity
1.1. B Cells and Autoantibodies in SLE
B lymphocytes are characterized by the expression on their membrane of the B-cell receptor (BCR). This receptor is physiologically devoted to the recognition of pathogens and the subsequent production of specific antibodies [28]. During the process of B-cell development, autoreactive B cells can also be generated. Although the development of these host-dangerous cells is controlled by immunological tolerance systems, such as clonal deletion or induction of peripheral anergy, these control mechanisms can sometimes fail. This allows the unwanted expansion and activation of such autoreactive B cells, with the possible onset of autoimmune diseases [29,30,31]. After development, B cells, including self-reactive B cells, require the intervention of soluble factors to ensure their survival and proliferation. Among these, the most important is the B-cell activating factor (BAFF), also known as B lymphocyte stimulator (BLys) [32,33]. The repertoire of autoantibodies produced by autoreactive B cells, targets mainly nuclear antigens. A key role in the generation of these autoantibodies is played by toll-like receptors (TLRs). Abnormal engagement of TLRs TLR7 and TLR9 subtypes in SLE, has been shown to effectively promote the production of autoantibodies against double-stranded DNA (dsDNA) and RNA-associated autoantigens, respectively [34,35,36,37]. Long-lived plasma cells (LLPCs) derived from the terminal differentiation of B cells, are an important source of autoantibody production in SLE. Short-lived plasmablasts, after interaction with CD4+ T cells in the germinal centers of the lymph nodes, have been shown to transform into high-affinity plasma cells that migrate to specific niches in the bone marrow, where they are protected from external events, being able to survive for a long time and continuing to produce autoantibodies [38]. Spontaneous formation of germinal centers, favoring the generation of LLPCs, is observed in both murine and human SLE, suggesting that this phenomenon is strictly involved in the genesis of autoantibody production [39]. Importantly, B lymphocytes may also play a role as antigen-presenting cells (APC) to autoreactive T lymphocytes in SLE, as demonstrated in mouse models [40,41]. A debated issue is the pathogenetic role of autoantibodies. Because the presence of autoantibodies can be detected in serum even years before the clinical signs of SLE, this has been considered an indication that these antibodies are a biomarker of the disease rather than a pathogenic factor. However, much evidence suggests their central role in the immunopathogenesis of SLE. Of particular importance, is the observation of the presence of immune complexes in lupus nephritis, at the glomerular level, formed by various autoantibodies, including anti-dsDNA antibodies, whose removal leads to amelioration of the disease [42,43,44]. Moreover, neonatal lupus erythematosus (NLE) develops as a result of the passive transfer of maternal autoantibodies across the placenta, which does not allow the passage of cells, including those of the immune system [45]. From these and other observations, it is possible to conclude that autoantibodies may contribute, at least to some extent, to the clinical manifestations of SLE.
1.2. T Cells in SLE Pathogenesis
Self-reactive T cells play a key role in the genesis of SLE. T-helper 1 (Th1) cells play a central role in the pathogenesis of SLE, by promoting oxidative stress related to IFNγ production [46]. In contrast, the number of IL-4-producing Th2 cells is reduced in the peripheral blood of SLE patients, suggesting their potential protective role, and that SLE activity may be associated with an increased IFNγ/IL-4 ratio [47]. T-helper 17 (Th17) cells are also involved in SLE pathogenesis. These cells are the main source of IL-17, a family of cytokines with potent inflammatory effects. In addition to their defensive action against pathogens, members of the IL-17 family can exacerbate tissue injury, because of their pro-inflammatory activity. IL-17 induces neutrophil recruitment, activation of the innate immune system, and enhancement of B-lymphocyte functions [48]. It has been reported that IL-17 levels correlate with SLEDAI in patients with LN [49,50]. Regulatory T cells (Tregs) are critical in maintaining peripheral tolerance to self-antigens. Although quantitative and qualitative differences in Tregs have been described in SLE, studies to date have shown conflicting results, and their role in SLE is still undefined. However, some studies have proposed that these cells, due to their ability to suppress effector T lymphocytes, could be considered in the basic cell therapy of SLE [51,52,53]. T-follicular helper (Tfh) cells are located in germinal centers and extrafollicular foci. These cells have been involved in the generation of autoreactive B-cell clones in murine and human SLE [54]. Tfh cells were found to aggregate in renal tissue with B cells, similar to what is observed in germinal centers in LN [55]. All these findings support the concept that interactions between CD4+ T cells and B cells are crucial in the development of autoimmunity, as they contribute decisively to the development and maintenance of autoreactive B cells and their differentiation into autoantibody-producing plasma cells. CD8+ T cells are also involved in the immunopathogenesis of SLE. Circulating CD8 T lymphocytes of SLE patients exhibit functional defects, including impaired cytolytic function, with reduced production of granzyme and perforin [56]. A depleted phenotype of circulating CD8 T lymphocytes in SLE patients has been associated with lower disease flare rates [57]. However, the qualitative abnormalities of CD8+ T lymphocytes are also related to the susceptibility of SLE patients to infections, which may be further exacerbated by the use of immunosuppressive drugs [58]. Finally, γδ-T lymphocytes were found in a higher percentage in SLE patients than in controls, suggesting their role in the autoimmune response [59,60].
2. The Role of Innate Immunity
2.1. Role of Neutrophils in SLE
It has been observed that in SLE, the neutrophil function is abnormal at several levels. First, neutrophils show reduced phagocytic capacity [61] and the inability to remove apoptotic cells, which are a known source of normally hidden self-antigens [62,63]. Variants of ITGAM, NCF1, and NCF2 genes, have been reported to be risk factors for SLE development, since they induce alteration of phagocytosis and dysregulate reactive oxide species production (ROS) [64,65,66]. It has also been reported that neutrophils can produce type-I IFN independently of TLRs stimulation and promote abnormal B-cell development in the bone marrow of SLE patients [67,68]. A subtype of neutrophils, called low-density granulocytes (LDG), is highly represented in the peripheral blood of SLE patients. These cells are associated with the presence of IFN signature and disease severity [69,70,71], and activate CD4+ T-cells to produce IFNγ and TNFα [69]. In SLE, LDGs are characterized by an increased ability to form neutrophil extracellular traps (NETs), released during their apoptosis (NETosis) [72]. NETs are rich in decondensed nucleic acids, and chromatin expelled outside the cells during the formation process can induce specific autoreactive immune responses against nucleic acid antigens [73]. Neutrophils are also characterized by the generation of ROS which, in normal conditions are responsible for cell killing, but in SLE contribute to endothelial damage [73]. Several genetic polymorphisms related to neutrophil dysregulation, that increase NET formation, have been described in SLE [74,75,76]. Moreover, neutrophils from SLE patients with mutations resulting in loss of STAT3 function, form NETs more spontaneously than healthy controls [77]. The increased formation of NETs and their reduced clearance may also lead to increased inflammasome activation in macrophages, amplifying the inflammatory response [78]. Taken together, these observations indicate that neutrophils, through NET formation, have very important immunostimulatory effects in SLE, contributing significantly to the immune dysregulation that leads to tissue damage.
2.2. Role of Plasmacytoid Dendritic Cells
Lymphoid-origin plasmacytoid dendritic cells (pDCs) are characterized by the ability to produce high levels of type-I IFN, thus playing a key role in the pathogenesis of SLE [79,80,81]. Production of type-I IFN occurs primarily in response to circulating ssRNA and dsDNA, that are internalized by pDCs through Fc-gamma receptor IIA (FcγRIIa). Nucleic acids are then recognized by TLR7 and TLR9 in the cytoplasm [82]. These receptors can also be activated by endogenous nucleic acids present in NETs. Once activated, TLRs trigger signaling pathways mainly involving the myeloid differentiation response gene-88 (Myd88) and interleukin-1 receptor-associated kinase 4 (IRAK4), leading to the activation of interferon regulatory factors 3 (IRF3) and IRF7 for IFN-I production [83,84,85]. The pDC-dependent production of type-I IFN is also important in linking innate and adaptive immunity. This occurs through complex interactions involving monocytes, neutrophils, natural killer cells, and T and B cells [86,87]. In this regard, the production of IFN-I by pDCs, can promote the differentiation of extrafollicular B cells into short-lived plasmablasts, which produce anti-dsDNA antibodies, thus creating a positive feedback loop supporting the autoimmune response, as demonstrated in animal models [88]. Activation of pDCs and high levels of IFN-I production, also increase the number and recruitment of pro-inflammatory T cells. This occurs particularly in the arterial wall. This finding has been associated with the development of accelerated atherosclerosis, as commonly observed in the course of SLE [89]. It should be noted, however, that pDCs may have also a tolerogenic function, through the generation of regulatory T cells (Treg). As mentioned earlier in this review, these cells are known to inhibit the activation of effector T cells [90,91,92]. Moreover, pDCs also facilitate the differentiation of immature B cells into IL-10-producing regulatory B cells (Breg), which can limit IFN-I production by pDCs, thus forming a regulatory feedback, the dysregulation of which is one of the most important components of SLE pathogenesis [93]. PDCs are therefore another possible target of SLE cell therapies. Figure 2 shows the relationships between the innate and adaptive immune systems in the pathogenesis of SLE.
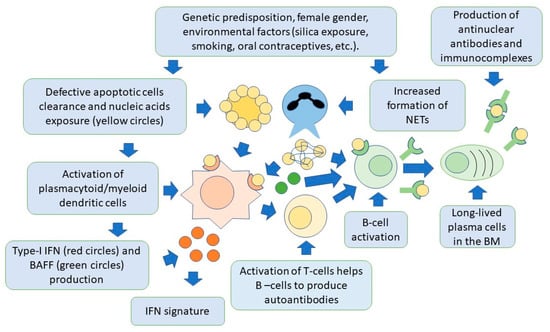
Figure 2. Both innate and adaptive immunity participate in the pathogenesis of SLE. In individuals with a genetic predisposition, and with the contribution of environmental factors, there is an accumulation of apoptotic cells and activation of NET production, by neutrophils. Cell nucleic acids are then exposed to the immune system and activate dendritic cells via toll-like receptors, to produce type-I IFN. This cytokine is responsible for activation of specific genes for pro-inflammatory factors by target cells (IFN signature). Dendritic cells also produce BAFF, which is necessary for B-cell activation and survival, and activate T cells, through the presentation of nuclear self-antigens. Self-reactive B cells are then activated to produce antibodies, and differentiate into long-lived plasma cells, localized in niches in the bone marrow, which are an additional source of autoantibodies. Autoantibodies form immunocomplexes with specific nuclear self-antigens that precipitate in tissues, contributing to organ damage.
3. The Role of Mitochondria
Mitochondria are organelles that provide the energy necessary for cell metabolism and survival, being the major source of adenosine triphosphate (ATP) synthesis [94]. These ancestral structures can release high quantities of mitochondrial DNA (mtDNA) after destruction, following cell apoptosis. MtDNA is extremely unstable, and its easy degradation can generate antigenic fragments [95]. MtDNA has been found to induce specific autoreactive T lymphocytes in patients with SLE [96,97]. These in turn may induce B cells to produce anti-DNA antibodies. It has also been observed that sequences of mtDNA are analogous to those of bacteria and therefore able to activate TLRs. TLR recognition triggers powerful downstream inflammatory responses, including type-I IFN production [98]. This pro-inflammatory response, further contributes to the breakdown of tolerance. It has also been reported that several mitochondrial gene variants are linked to the risk of developing SLE, as demonstrated in mouse models [99,100,101]. Mitochondrial polymorphisms increase oxidative stress [102], as demonstrated by the accumulation of oxidized nucleic acids in the mitochondria of neutrophils of patients with SLE [103]. The accumulated oxidized mtDNA can be therefore extruded during NETosis, potentially triggering type-I IFN activation by plasmacytoid dendritic cells (pDCs) [63]. Finally, mitochondrial RNA (mtRNA) is another source of autoantigens, and titers of RNA autoantibodies against mtRNA are significantly higher in patients with SLE compared with controls [104].
4. The Role of Apoptosis
It has recently been shown that enzymes such as nucleases are critical for nucleic acid digestion and maintenance of tolerance [105]. A deficiency of nucleases is responsible for lupus-like manifestations in mouse models [106]. Recent studies have identified neutralizing antibodies against DNASE1L3 in some SLE patients, resulting in the accumulation of extracellular DNA and the formation of immune complexes [107]. Mice KO for DNaseII genes does not survive due to undigested DNA in phagocytes [108,109]. Mice KO for genes encoding TREX1 nuclease develops lupus-like symptoms, including skin lesions, vasculitis, nephritis, and sometimes systemic inflammation [110,111]. These observations lead to the conclusion that undigested nucleosomes are an initial inducer of the autoimmune responses, as observed in SLE patients. Apoptotic cell clearance can induce the breakdown of immune tolerance through several mechanisms. These include the activation of signals mediated by pattern recognition receptors (PRRS) [112,113,114]. The role of apoptosis defects in SLE has been confirmed in human studies, showing that patients with SLE have a defect in apoptotic cell clearance [115,116]. However, it should be noted that, in some mouse models with impaired apoptosis, autoimmunity does not occur, indicating that other events are required to induce the onset of SLE [117]. NETs are also involved in the apoptosis deficit in SLE. The molecular components of NETs can be complexed with DNA, making it resistant to enzymatic digestion by DNases, inducing type-I IFN production by plasmacytoid dendritic cells [118].
5. The Role of Interferons in SLE
A recent finding on the pathogenesis of SLE that opened new lines of research for innovative drug development, was the recognition of a high type-I IFN signature in SLE patients [119]. IFNs play a pivotal role in defense against pathogens. Three types of IFNs can be distinguished: type-I IFN, which is a family composed of IFNα (13 subtypes), IFNβ, IFNω, IFNκ, and IFNε; type-II IFN, also known as IFNγ; and type-III IFN (IFNλ) [3,120,121]. Type-I IFN, particularly the IFNα and IFNβ family members, is the one mainly involved in the pathogenesis of SLE. Type-I IFN is induced by the activation of pathogen recognition receptors (PRRs) such as toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I), and melanoma differentiation-associated protein 5 (MDA5). All these molecules are activated by nucleic acids or by bacterial products such as lipopolysaccharides and peptidoglycan [122]. Although virtually any cell type can produce type-I IFN [123], very high levels of this cytokine are synthesized by pDCs, as already discussed [124,125,126]. Type-I IFNs recognize the IFNα receptor (IFNAR), a heterodimeric complex that in turn activates intracellular signaling through Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2). These proteins phosphorylate transcriptional signal transducers and activators STAT 1 and STAT 2. These intracellular molecules bind IRF7 and IRF9, to form the ISGF3 complex. This complex translocates into the nucleus, where it induces transcription of genes named IFN-sensitive response elements (ISREs), encoding for proteins contributing to the inflammatory cascade [127,128]. Initial experimental animal model studies showed that the administration of type-I IFN was able to induce the production of autoantibodies and contribute to organ damage [129]. The earliest evidence suggesting that type one interferon could play a key role in the genesis of SLE in humans, came from the observation that patients treated with IFNα for hepatitis C [130] or neoplastic disease [131,132], could develop antinuclear antibody positivity and in some cases lupus-like syndromes. These clinical conditions regressed after discontinuation of IFNα treatment [133,134]. It has been shown that polymorphisms in genes along the type-I IFN signaling pathways represent important genetic risk factors for the occurrence of SLE-including IRF genes [135,136,137]. Polymorphisms have been also described in STAT4, STAT3, and TYK2 genes [77,138]. The IFN signature is also emerging as a possible biomarker for precision treatment with novel anti-IFN therapeutic agents, as discussed in more detail in the section on SLE therapy.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24076578
This entry is offline, you can click here to edit this entry!