Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Pulmonary thrombosis in situ is a pathological condition nonrelated to embolism from deep vein thrombosis (DVT) in the lower extremities.
- COVID-19
- SARS-CoV-2 infection
- pulmonary in situ thrombosis
- immunothrombosis
1. Introduction
COVID-19, the infectious disease caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), is frequently associated with micro- and macrovascular thrombotic events, which are associated with disease severity and worse clinical outcomes [1][2][3]. Severe hypercoagulability mainly develops in the lungs of COVID-19 patients [4], and the most common vascular thrombotic complication involves the pulmonary arteries [1][5][6]. Post mortem lungs from patients with SARS-CoV-2 infection showed severe coagulation abnormalities, especially fibrin- and platelet-rich thrombi, not observed in non-COVID-19 autopsy controls [4]. The “cytokine storm” and its associated diffuse endothelial dysfunction, common in severe SARS-CoV-2 infection, could also increase the risk of developing nonrespiratory complications, such as neurological involvement, preeclampsia, and orchitis-like syndromes [7][8][9]. COVID-19-associated thrombotic complications are secondary to a synergistic interplay of endotheliopathy, coagulation pathways, platelet dysfunction, and detrimental immune-mediated thrombosis [10]. However, it is not clear which of the pathobiological mechanisms, conventional risk factors, and venous thromboembolic disease, in situ immunothrombosis, or additional thrombotic mechanisms contributes more to these COVID-19-associated pulmonary thrombotic events [2][11].
2. COVID-19-Associated Pulmonary Thrombosis Is an In Situ Immunothrombosis
Pulmonary thrombosis in situ is a pathological condition nonrelated to embolism from deep vein thrombosis (DVT) in the lower extremities [12]. Nonpulmonary in situ thrombosis in the right ventricle was also described in COVID-19 patients [13][14]. COVID-19-associated pulmonary intravascular coagulopathy is a complex disease “orchestrated” by a severe and dysregulated proinflammatory response that can lead to immunothrombosis [2][15]. The prothrombotic state in patients with COVID-19 is reminiscent of this immunothrombosis process, a result of the crosstalk between the immune and hemostatic systems and characterized by the production of microthrombi in small capillaries, in which endothelial cells (ECs) adopt a proadhesive phenotype in contact with SARS-CoV-2 [16][17][18]. Pathological studies showed multiple microthrombi in pulmonary capillaries and larger primary thrombi in arterioles considered to be primary in nature [14]. Autopsy lung samples from patients with COVID-19 showed important circulatory changes with inflammation-dependent intravascular thrombosis, direct pathological evidence for immunothrombosis, which were not found in other organs such as heart, brain, and kidneys [14]. There is a concept of a local lung-associated coagulation system, the “bronchoalveolar hemostasis” and the formation of blood clots in the microvasculature of the lungs could be a part of the host immune defense against SARS-CoV-2 [19][20]. Immunothrombosis is an interplay between systemic and lung inflammatory pathways and coagulation/fibrinolysis systems (Figure 1).
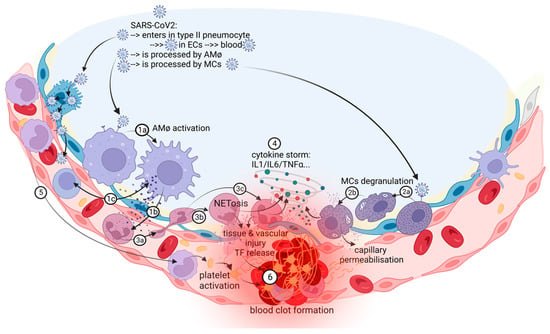
Figure 1. The in situ immunothrombosis in COVID-19. SARS-CoV-2 replicates into the type II pneumocytes. Furthermore, SARS-CoV-2 could be transferred to adjacent ECs that have ACE-2 receptors and passed into the blood. AMφs interact with SARS-CoV-2 and activate themselves (1a). After activation, the major process in COVID-19 is the chemoattraction of neutrophils (1b). AMφs could also act like antigen presenters to T lymphocytes (1c). Neutrophils, attracted by AMφs, come into the area of the immune conflict, crawling, and squeezing (3a), and they activate and release neutrophil extracellular traps (NETosis) (3b); aberrant NETosis and oxidants released (3c) into the environment contribute to the “cytokine storm” (4), tissue and vascular injuries and initiate immune-activated coagulation (6). SARS-CoV-2 activates MCs (2a) and they release histamine, other specific proteases, prostaglandins and leukotrienes, cytokines and chemokines (2b), contributing to capillary permeabilization and to the “cytokine storm” (4), ending with in situ immunothrombosis (6). Monocytes are excessively recruited in the area (5) and contribute to the “cytokine storm” (4) and to the activation of coagulation pathways (6). The blood clot (6) contains RBCs, activated platelets, fibrin, NETs, and lymphocytes supporting the immune mechanism for thrombosis.
In patients with in situ pulmonary thrombosis, in-depth immune pathology analysis by immunohistochemistry supports the inflammatory nature of arterial thrombi composed of white blood cells, especially neutrophils, CD3+ and CD20+ lymphocytes, fibrin, red blood cells, and platelets, but not megakaryocytes [15]. Regional thrombosis of the lung microvasculature also showed compressed deformed red blood cells (RBCs), including polyhedrocytes and different morphological types of fibrin structures coated with sparse spherical microparticles, which could comprise virions or cellular ectosomes [14]. Different phenotypes of in situ thrombosis may exist and need different intensity of anticoagulant therapy [21].
Severe COVID-19 is characterized by a proinflammatory state and an associated disbalance in hemostasis, which starts with the disruption of the alveolar epithelium, and involves intrinsic and extrinsic coagulation pathways, neutrophil extracellular traps (NETs) activation and release (NETosis), and impaired fibrinolysis secondary to high plasminogen activator inhibitor 1 (PAI-1) levels [17]. Acute respiratory failure in patients with severe COVID-19 is associated with diffuse alveolar damage, perialveolar microangiopathy, and obstructive neoangiogenesis [17][22][23][24][25][26][27][28][29]. In situ pulmonary thrombosis may appear in COVID-19 pneumonia patients, with peripheral distribution, either within the consolidation lesions of the infected lungs (due to active local inflammation), or in nonconsolidation areas (due to hypercoagulability caused by systemic inflammation) [21]. Systemic inflammatory markers such as C-reactive protein (CRP) have higher values in patients with COVID-19-associated pulmonary artery thrombosis compared with those without pulmonary thrombi [30][31][32][33]. Inflammatory-mediated thrombosis is also supported by the similarity in terms of comorbidities and other classical risk factors for venous thromboembolic disease in patients with and without COVID-19-associated pulmonary thrombosis [30][31]. Elevated serum levels of CRP are associated with thrombotic disease and mortality in COVID-19 patients [34][35][36][37]. Moreover, CRP is an important link between inflammation and thrombosis, as it can activate the complement cascade, induce platelet adhesion to ECs, stimulate tissue factor (TF) expression by blood monocytes, and alter the fibrinolytic balance of ECs [38]. Ferritin, another inflammatory biomarker, is also an independent predictive factor for both venous thrombotic events, especially pulmonary thrombosis, and also mortality, in COVID-19 patients [6][39][40]. Ferritin induces mitochondrial dysfunction in platelets, and this could also contribute to inflammation and thrombosis [2].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11030929
References
- Manolis, A.S.; Manolis, T.A.; Manolis, A.A.; Papatheou, D.; Melita, H. COVID-19 Infection: Viral Macro- and Micro-Vascular Coagulopathy and Thromboembolism/Prophylactic and Therapeutic Management. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 12–24.
- Loo, J.; Spittle, D.A.; Newnham, M. COVID-19, immunothrombosis and venous thromboembolism: Biological mechanisms. Thorax 2021, 76, 412–420.
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847.
- Won, T.; Wood, M.K.; Hughes, D.M.; Talor, M.V.; Ma, Z.; Schneider, J.; Skinner, J.T.; Asady, B.; Goerlich, E.; Halushka, M.K.; et al. Endothelial thrombomodulin downregulation caused by hypoxia contributes to severe infiltration and coagulopathy in COVID-19 patient lungs. eBioMedicine 2022, 75, 103812.
- Mueller-Peltzer, K.; Krauss, T.; Benndorf, M.; Lang, C.N.; Bamberg, F.; Bode, C.; Duerschmied, D.; Staudacher, D.L.; Zotzmann, V. Pulmonary artery thrombi are co-located with opacifications in SARS-CoV2 induced ARDS. Respir. Med. 2020, 172, 106135.
- Oba, S.; Hosoya, T.; Amamiya, M.; Mitsumura, T.; Kawata, D.; Sasaki, H.; Kamiya, M.; Yamamoto, A.; Ando, T.; Shimada, S.; et al. Arterial and Venous Thrombosis Complicated in COVID-19: A Retrospective Single Center Analysis in Japan. Front. Cardiovasc. Med. 2021, 8, 767074.
- Delli Muti, N.; Finocchi, F.; Tossetta, G.; Salvio, G.; Cutini, M.; Marzioni, D.; Balercia, G. Could SARS-CoV-2 infection affect male fertility and sexuality? Apmis 2022, 130, 243–252.
- Marshall, M. How COVID-19 can damage the brain. Nature 2020, 585, 342–343.
- Tossetta, G.; Fantone, S.; Delli Muti, N.; Balercia, G.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Preeclampsia and severe acute respiratory syndrome coronavirus 2 infection: A systematic review. J. Hypertens. 2022, 40, 1629–1638.
- Portier, I.; Campbell, R.A.; Denorme, F. Mechanisms of immunothrombosis in COVID-19. Curr. Opin. Hematol. 2021, 28, 445–453.
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649.
- Payus, A.O.; Lin, C.L.S.; Ibrahim, A. The poorly understood yet potent risk of pulmonary artery thrombosis in-situ in Post-Acute COVID-19 syndrome. J. Cardiothorac. Surg. 2023, 18, 42.
- Cuevas Vilaplana, A.; Roldan Torres, I.; Vizuete Del Rio, J. Myocarditis and in situ thrombosis in the right ventricle in a COVID-19 patient. Hipertens. Riesgo Vasc. 2021, 38, 148–150.
- Khismatullin, R.R.; Ponomareva, A.A.; Nagaswami, C.; Ivaeva, R.A.; Montone, K.T.; Weisel, J.W.; Litvinov, R.I. Pathology of lung-specific thrombosis and inflammation in COVID-19. J. Thromb. Haemost. 2021, 19, 3062–3072.
- Quartuccio, L.; Sonaglia, A.; Casarotto, L.; McGonagle, D.; Di Loreto, C.; Pegolo, E. Clinical, laboratory and immunohistochemical characterization of in situ pulmonary arterial thrombosis in fatal COVID-19. Thromb. Res. 2022, 219, 95–101.
- Agrati, C.; Sacchi, A.; Tartaglia, E.; Vergori, A.; Gagliardini, R.; Scarabello, A.; Bibas, M. The Role of P-Selectin in COVID-19 Coagulopathy: An Updated Review. Int. J. Mol. Sci. 2021, 22, 7942.
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284.
- Lim, M.S.; McRae, S. COVID-19 and immunothrombosis: Pathophysiology and therapeutic implications. Crit. Rev. Oncol. Hematol. 2021, 168, 103529.
- Thachil, J.; Srivastava, A. SARS-2 Coronavirus-Associated Hemostatic Lung Abnormality in COVID-19: Is It Pulmonary Thrombosis or Pulmonary Embolism? Semin. Thromb. Hemost. 2020, 46, 777–780.
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767.
- Wang, L.; Chen, F.; Bai, L.; Yi, Q.; Peng, Y. In situ pulmonary thrombosis in patients with COVID-19 pneumonia: Different phenotypes may exist. Thromb. Res. 2020, 196, 541–542.
- Martín Giménez, V.M.; Inserra, F.; Tajer, C.D.; Mariani, J.; Ferder, L.; Reiter, R.J.; Manucha, W. Lungs as target of COVID-19 infection: Protective common molecular mechanisms of vitamin D and melatonin as a new potential synergistic treatment. Life Sci. 2020, 254, 117808.
- Calabrese, F.; Pezzuto, F.; Fortarezza, F.; Hofman, P.; Kern, I.; Panizo, A.; von der Thüsen, J.; Timofeev, S.; Gorkiewicz, G.; Lunardi, F. Pulmonary pathology and COVID-19: Lessons from autopsy. The experience of European Pulmonary Pathologists. Virchows Arch. 2020, 477, 359–372.
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128.
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209.
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140.
- Duarte-Neto, A.N.; Monteiro, R.A.A.; da Silva, L.F.F.; Malheiros, D.; de Oliveira, E.P.; Theodoro-Filho, J.; Pinho, J.R.R.; Gomes-Gouvêa, M.S.; Salles, A.P.M.; de Oliveira, I.R.S.; et al. Pulmonary and systemic involvement in COVID-19 patients assessed with ultrasound-guided minimally invasive autopsy. Histopathology 2020, 77, 186–197.
- Cacciola, R.; Gentilini Cacciola, E.; Vecchio, V.; Cacciola, E. Cellular and molecular mechanisms in COVID-19 coagulopathy: Role of inflammation and endotheliopathy. J. Thromb. Thrombolysis 2022, 53, 282–290.
- Golubeva, M.G. Role of P-Selectin in the Development of Hemostasis Disorders in COVID-19. Biol. Bull. Rev. 2022, 12, 406–413.
- Vivan, M.A.; Rigatti, B.; da Cunha, S.V.; Frison, G.C.; Antoniazzi, L.Q.; de Oliveira, P.H.K.; Oliveira, J.P.S.; Fontanari, C.; Seligman, B.G.S.; Seligman, R. Pulmonary embolism in patients with COVID-19 and D-dimer diagnostic value: A retrospective study. Braz. J. Infect. Dis. 2022, 26, 102702.
- Močibob, L.; Šušak, F.; Šitum, M.; Višković, K.; Papić, N.; Vince, A. COVID-19 and Pulmonary Thrombosis-An Unresolved Clinical Puzzle: A Single-Center Cohort Study. J. Clin. Med. 2022, 11, 7049.
- Jalde, F.C.; Beckman, M.O.; Svensson, A.M.; Bell, M.; Skold, M.; Strand, F.; Nyren, S.; Kistner, A. Widespread Parenchymal Abnormalities and Pulmonary Embolism on Contrast-Enhanced CT Predict Disease Severity and Mortality in Hospitalized COVID-19 Patients. Front. Med. 2021, 8, 666723.
- Masselli, G.; Almberger, M.; Tortora, A.; Capoccia, L.; Dolciami, M.; D’Aprile, M.R.; Valentini, C.; Avventurieri, G.; Bracci, S.; Ricci, P. Role of CT angiography in detecting acute pulmonary embolism associated with COVID-19 pneumonia. Radiol. Med. 2021, 126, 1553–1560.
- Smilowitz, N.R.; Kunichoff, D.; Garshick, M.; Shah, B.; Pillinger, M.; Hochman, J.S.; Berger, J.S. C-reactive protein and clinical outcomes in patients with COVID-19. Eur. Heart J. 2021, 42, 2270–2279.
- Sultana, G.N.N.; Srivastava, A.; Akhtaar, K.; Singh, P.P.; Islam, M.A.; Mishra, R.K.; Chaubey, G. Studying C-reactive protein and D-dimer levels in blood may prevent severe complications: A study in Bangladeshi COVID-19 patients. Front. Genet. 2022, 13, 966595.
- Gorog, D.A.; Storey, R.F.; Gurbel, P.A.; Tantry, U.S.; Berger, J.S.; Chan, M.Y.; Duerschmied, D.; Smyth, S.S.; Parker, W.A.E.; Ajjan, R.A.; et al. Current and novel biomarkers of thrombotic risk in COVID-19: A Consensus Statement from the International COVID-19 Thrombosis Biomarkers Colloquium. Nat. Rev. Cardiol. 2022, 19, 475–495.
- Lucijanić, M.; Stojić, J.; Atić, A.; Čikara, T.; Osmani, B.; Barišić-Jaman, M.; Andrilović, A.; Bistrović, P.; Zrilić Vrkljan, A.; Lagančić, M.; et al. Clinical and prognostic significance of C-reactive protein to albumin ratio in hospitalized coronavirus disease 2019 (COVID-19) patients. Wien. Klin. Wochenschr. 2022, 134, 377–384.
- Fay, W.P. Linking inflammation and thrombosis: Role of C-reactive protein. World J. Cardiol. 2010, 2, 365–369.
- Galland, J.; Thoreau, B.; Delrue, M.; Neuwirth, M.; Stepanian, A.; Chauvin, A.; Dellal, A.; Nallet, O.; Roriz, M.; Devaux, M.; et al. White blood count, D-dimers, and ferritin levels as predictive factors of pulmonary embolism suspected upon admission in noncritically ill COVID-19 patients: The French multicenter CLOTVID retrospective study. Eur. J. Haematol. 2021, 107, 190–201.
- Kaushal, K.; Kaur, H.; Sarma, P.; Bhattacharyya, A.; Sharma, D.J.; Prajapat, M.; Pathak, M.; Kothari, A.; Kumar, S.; Rana, S.; et al. Serum ferritin as a predictive biomarker in COVID-19. A systematic review, meta-analysis and meta-regression analysis. J. Crit. Care 2022, 67, 172–181.
This entry is offline, you can click here to edit this entry!