Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Regulatory T cells (Tregs) and T helper 17 cells (Th17) are two CD4+ T cell subsets with antagonist effects. Th17 cells promote inflammation, whereas Tregs are crucial in maintaining immune homeostasis. Th17 cells and Treg cells are the foremost players in several inflammatory diseases.
- Th17 cells
- Treg cells
- inflammation
- lung
1. Introduction
The immune system acts as the guardian of the host and functions to defend against foreign antigens, induce self-tolerance, and promote immunological memory. However, it is not protective or beneficial all the time. The individual’s tissue components may be attacked by the immunological reaction resulting in autoimmune diseases in specific settings. It is certain that a single theory or mechanism cannot explain autoimmune diseases. As proposed by Shoenfeld and Isenberg, autoimmune diseases are caused by various factors, including immunological, genetic, hormonal, and environmental factors [1]. Non-genetic components rather than inherent components play a dominant role in determining disease susceptibility and severity, which has been demonstrated by the discordance of autoimmune diseases in identical twins [2]. Immunological factors play a vital role in the initiation, progression, and regression of autoimmune diseases. In a typical setting, T cells are tolerant to physiological levels of self-antigen. However, this state of tolerance breaks down in some individuals, resulting in autoimmune/inflammatory diseases. One of the critical features of inflammatory diseases is the deregulated Th1/T helper 17 cells (Th17) responses, frequently accompanied by a reduction and/or alteration of regulatory T (Treg) cells. Th17 cells serve as inflammatory cells, which in excess, promote inflammatory diseases. On the other hand, Treg cells show suppressor function, which, when in failure, contributes to the same disease [3].
1.1. Th17 Cells
Initial studies by Infante-Duarte et al. identified CD4+ T cells producing IL-17A as a T helper cell subset distinct from Th1 and Th2 cell subsets [4]. This subset, called Th17 cells, predominantly produces interleukin-17A (IL-17A), IL-17F, IL-21, and IL-22 [5]. IL-17A, originally named CTLA8, was cloned and described by Rouvier et al. [6]. It is a homodimeric glycoprotein with 155 amino acids linked by disulfide bonds. IL-17F, also produced by Th17 cells, shows 55% similarity with IL-17A, and they form IL-17F homodimers, IL-17A homodimers, or IL-17A-IL-17F heterodimers [7]. IL-17 binds to its receptor (IL-17R), a transmembrane protein, highly expressed in rats and mice’s spleen, kidneys, liver, and lungs [8]. Th17 cells require the transcription factor, RORγt, and cytokine IL-6 in combination with transforming growth factor-β (TGF-β) for their differentiation [9]. IL-6 acts as a major factor guiding the differentiation of T cells into Th17 cells or Treg cells [9]. IL-21, together with TGF-β, also functions as an alternative pathway to generate Th17 cells [10]. Once they reach the site of inflammation, IL-17 released by Th17 cells stimulates the expression of pro-inflammatory cytokines like granulocyte-macrophage colony-stimulating factor, Granulocyte-colony stimulating factor, IL-6 and tumor necrosis factor-alpha (TNF-α) [11]. In addition, IL-17 also promotes the secretion of CXC chemokines, which attracts neutrophils in vivo [11]. Moreover, IL-17 stimulates the production of antimicrobial peptides, such as β-defensin and S100 proteins, providing defense against a wide range of microorganisms [12][13].
1.2. Treg Cells
As the bias towards pro-inflammatory cytokines and cells induces the development and perpetuation of autoimmunity, immunoregulatory factors are thought to straighten out the laterality. Regulatory T cells are crucial members of the family of immunoregulatory cells that preserve self-tolerance and fine-tune the immune response. Treg cells suppress inflammation by cell-cell contact or releasing cytokines, such as IL-10 or TGF-β, and they require the transcription factor FoxP3 for their differentiation [3][14]. In recent years, research has identified two types of Treg cells called natural Treg cells (nTreg) and inducible Treg cells (iTreg). nTreg cells develop in the thymus, and when entering peripheral tissues, they suppress self-reactive T cells. Studies in both mice and humans found that nTreg cells constitute around 10% of CD4 T cells in the periphery [15]. They express FoxP3 before they are released from the thymus, and the expression of TGF-β helps in their maintenance of inhibitory function after they migrate from the thymus [3][14]. Inducible Treg cells develop from naive T cells in the secondary lymphoid organ upon antigen exposure. Following interaction with TCR, TGF-β induce the FoxP3 expression in CD4+ CD25− cells, thereby, converting them to FoxP3+ CD4+ CD25+ cells [16]. These iTreg cells mediate their inhibitory activities by secretion of IL-10 or TGF-β, which is crucial for inhibiting overexuberant immune response [17] (Figure 1).
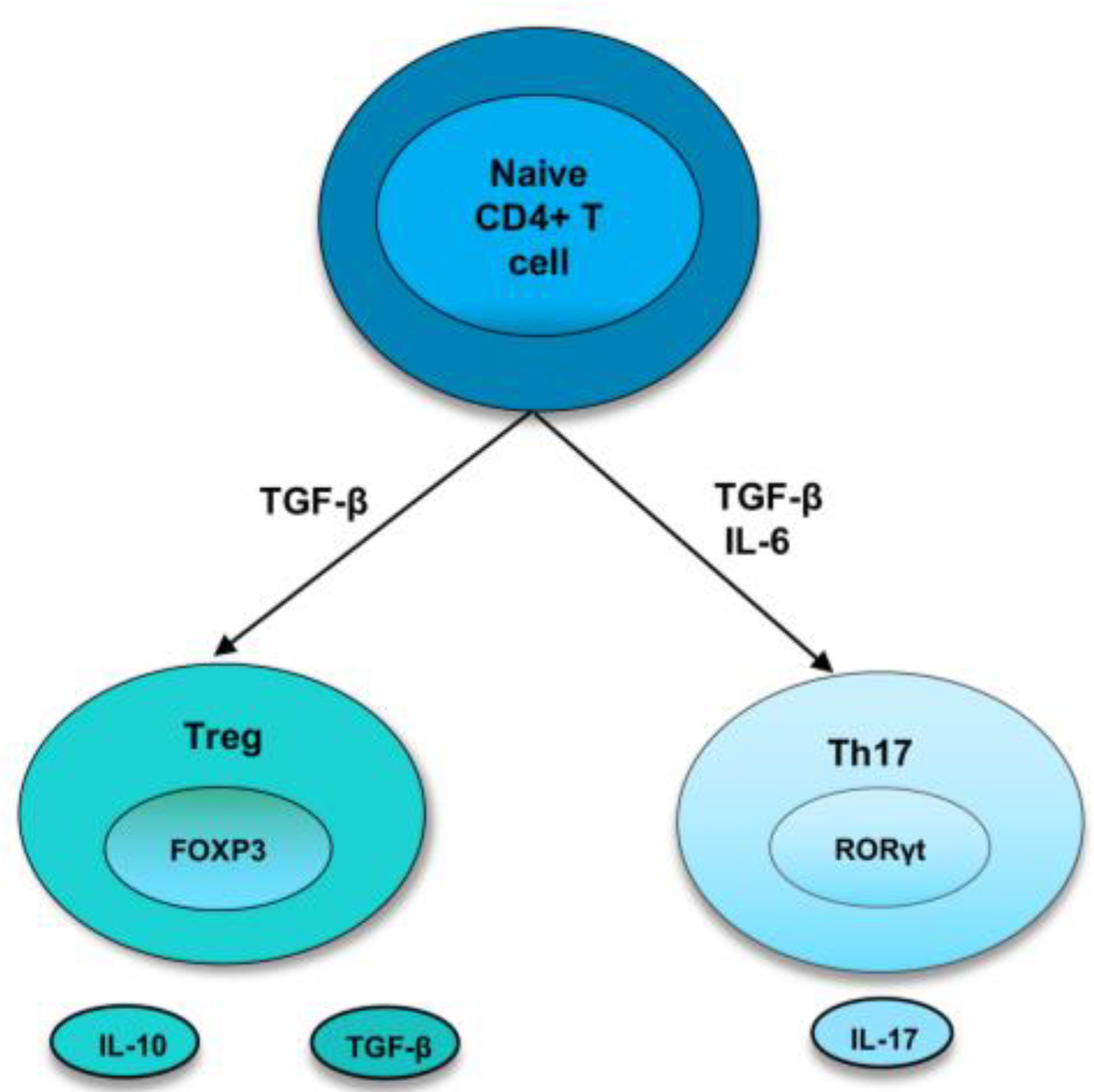
Figure 1. Differentiation of naive T cells into Th17 and Treg cells. In naive CD4+ T cells, TGF-β induce the development of Tregs by promoting Foxp3 expression. Treg cells express cytokines, IL-10 and TGF-β. However, in the presence of IL-6 and TGFβ, RORγt is induced, leading to a Th17 phenotype.
2. Th17/Treg Cells in Lung Inflammatory Diseases
2.1. Chronic Obstructive Pulmonary Disease (COPD)
COPD is a chronic inflammatory lung disease characterized by airway and/or alveolar abnormalities that cause obstructed airflow from the lungs [18]. Studies over the last decade highlighted the relevance of maintaining the balance between Th17 cells and Treg cells to control the inflammatory response in COPD. An increased Th17 response is involved in the progression of Chronic Obstructive Pulmonary Disease (COPD) in both clinical and experimental studies [18]. Th17 cytokine, IL-17A, levels were higher in the sputum of patients with COPD stages 3 and 4 compared to non-smokers and healthy smokers [19]. Reduced numbers of Treg cells were observed in the bronchial epithelium of severe/very severe COPD patients than in those with mild and moderate COPD and healthy smokers [20].
2.2. Acute Respiratory Distress Syndrome (ARDS)
Acute respiratory distress syndrome (ARDS) is an important cause of acute pulmonary failure with severe disease and mortality [21]. The most common cause of ARDS is bacterial or viral pneumonia [21]. ARDS is characterized by dysregulated inflammation, increased permeability of microvascular barriers, and uncontrolled activation of coagulation pathways [21]. Activation of several immune cells, including neutrophils, macrophages, and dendritic cells, plays an important role in the development of ARDS [22]. The involvement of CD4+ T cells has been revealed recently for the pathogenesis of ARDS. ARDS patients show a higher frequency of Th17 cells and IL-17 compared to the control group [22]. The Th17/Treg ratio is higher in the peripheral blood of ARDS patients compared with the healthy controls [22]. A higher Th17/Treg ratio is associated with more adverse outcomes in ARDS patients. Mechanistically, recent studies demonstrated that secreted phosphoprotein 1 (SPP1) exacerbated lung inflammation in ARDS by modulating Th17/Treg balance [23]. SPP1 reduces the ubiquitination and degradation of HIF-1α, which, in turn, leads to a higher Th1/Treg ratio. IL-33 production in LPS-induced ARDS is reported to increase the Th17/Treg ratio [24]. IL-33 deficiency inhibits the differentiation of T cells into Th17 cells and restores Th17/Treg balance. Consequently, IL-33 deficiency significantly reduces inflammation in LPS-induced ARDS, whereas recombinant IL-33 treatment exacerbates lung inflammation [24].
2.3. Sarcoidosis
Sarcoidosis is an inflammatory disorder characterized by granulomatous inflammation that affects multiple organs, mostly the lungs and mediastinal lymph nodes [25]. Emerging studies suggest the pleiotropic functions of Th17 and Treg cells in the pathogenesis of sarcoidosis. Higher IL-17A cytokine production is observed in the BALF of patients with pulmonary sarcoidosis [25]. Moreover, a higher Th17/Treg ratio was observed in peripheral blood and BAL of patients with active and progressive sarcoidosis [26]. After treatment with corticosteroids, the level of Foxp3 mRNA was elevated in the peripheral blood, and expression of RORγt mRNA was reduced [26].
2.4. Asthma
Asthma is a chronic inflammatory disease of the airways involving inflammatory cells such as mast cells, eosinophils, neutrophils, macrophages, and T lymphocytes [27]. Typically, asthmatic inflammation is mediated by excessively activated Th2 cells eosinophilia [27], but recent studies showed the involvement of cytokine IL-17A in multiple asthma pathogenesis, including neutrophilic inflammation, steroid insensitivity, activation of epithelial cells, and airway remodeling [28]. A large number of cells positive for IL-17 are reported in the sputum and bronchioalveolar fluids (BALFs) of asthmatic patients [29]. In addition, many reports identified that levels of IL-17A are correlated positively with the severity of asthma [30][31][32]. Inhibition of IL-17 in a model of LPS-induced asthma exacerbation aid in controlling Th2 and Th17 responses and signaling pathways associated with inflammation and remodeling [33].
2.5. Pulmonary Infectious Diseases
In addition to their role in non-infectious inflammatory lung diseases, maintaining Th17 /Treg balance is important for protective immunity against lung infections. Human IL-17A and IL-17F are crucial for protective immunity against mucocutaneous candidiasis [34]. Treg cells prevent the differentiation of naïve T cells into Th17 cells and prevent the clearance of Candida albicans infection [35]. IL-17 is identified as a critical factor required for protective immunity to Pneumocystis infection. Administration of anti-IL-17 neutralizing antibody to wild-type mice infected with P. carinii resulted in severe fungal infection [36]. Similarly, regulatory T cells are recruited to the lung during the course of Pneumocystis infection in mice [37]. Depletion of the Treg population results in increased levels of IL-1β and IL-6, leading to increased lung injury [37]. Th17/Treg balance also acts as a critical factor for controlling lung inflammation during chlamydial infection. IL-17A produced by Th17 cells during chlamydial lung infection has a significant impact on the development of protective type 1 immunity [38][39][40]. Chlamydial lung infection of mice induced IL-17 production in lung and lymph nodes at earlier and later stages of infection [41]. Neutralization of IL-17 in mice resulted in higher body weight loss, bacterial burden, and more severe pathological changes in the lung compared with sham-treated control mice [41]. IL-17 neutralized mice exhibit reduced Th1 responses, increased Th2 responses, and altered DC phenotype. Moreover, the adoptive transfer of DC isolated from IL-17-neutralized mice failed to protect the recipients against challenge infection compared to DC from sham-treated mice [41].
On the other hand, higher Treg responses contribute to tissue pathology after chlamydial lung infection [39][40]. Treg cells are observed in the chlamydial infection site of both humans and mice [42][43][44]. Recent studies suggested that NK cells provide protective immunity to chlamydial lung infection by maintaining Th17/Treg balance [45][46]. During Chlamydophila pneumoniae (Cpn) lung infection, NK cell depletion increased the number of Treg cells and IL-10-producing CD4+ T cells. The changes in T cell responses were associated with severe disease and bacterial load in the lung. Adoptive transfer of DCs from NK cell-deficient mice induced Treg cells in the recipient mice, which promotes pathological response [45]. In the mice model of Chlamydia muridarum lung infection, NK cell depletion resulted in lower IL-17 cytokine production and Th17 cells [46].
3. Conclusions
The importance of the balance between pro-inflammatory and anti-inflammatory cytokines and cells in maintaining immune homeostasis is widely acknowledged. Th17 cells promote inflammation and pathology, whereas Treg cells maintain self-tolerance. The balance between inflammation and self-tolerance is disrupted, leading to inflammation.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24054865
References
- Shoenfeld, Y.; Isenberg, D. The mosaic of autoimmunity. Immunol. Today 1989, 10, 123–126.
- Javierre, B.M.; Fernandez, A.F.; Richter, J. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010, 20, 170–179.
- Noack, M.; Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014, 13, 668–677.
- Infante-Duarte, C.; Horton, H.F.; Byrne, M.C.; Kamradt, T. Microbial Lipopeptides Induce the Production of IL-17 in Th Cells. J. Immunol. 2000, 165, 6107–6115.
- Ouyang, W.; Kolls, J.K.; Zheng, Y. The Biological Functions of T Helper 17 Cell Effector Cytokines in Inflammation. Immunity 2008, 28, 454–467.
- Rouvier, E.; Luciani, M.F.; Mattéi, M.G.; Denizot, F.; Golstein, P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J. Immunol. 1993, 150, 5445–5456.
- Andrew, J.L.; Frann, B.; Matthew, J.W.; Riyez, K.; Mary, C.; Samuel, J.G.; Kyriaki, D.-J.; Cara MMWJill, F.W.; Lynette, A.F. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J. Immunol. 2007, 179, 7791–7799.
- Yao, Z.; Fanslow, W.C.; Seldin, M.F.; Rousseau, A.-M.; Painter, S.L.; Comeau, M.R.; Cohen, J.I.; Spriggs, M.K. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity 1995, 3, 811–821.
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238.
- Korn, T.; Bettelli, E.; Gao, W.; Awasthi, A.; Jäger, A.; Strom, T.B.; Kuchroo, V.K. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007, 448, 484–487.
- Xu, S.; Cao, X. Interleukin-17 and its expanding biological functions. Cell Mol. Immunol. 2010, 7, 164–174.
- Kao, C.Y.; Yin, C.; Philip, T.; Shinichiro, W.; Fei, H.; Christy, K.; Richart, W.H.; Reen, W. IL-17 markedly up-regulates beta-defensin-2 expression in human airway epithelium via JAK and NF-kappaB signaling pathways. J. Immunol. 2004, 173, 3482–3491.
- Ganz, T. Defensins and Host Defense. Science 1999, 286, 420–421.
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336.
- Wan, Y.Y.; Flavell, R.A. TGF-beta and regulatory T cell in immunity and autoimmunity. J. Clin. Immunol. 2008, 28, 647–659.
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; Wahl, S.M. Conversion of peripheral CD4+ CD25− naive T cells to CD4+ CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886.
- Rubtsov, Y.P.; Rudensky, A.Y. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat. Rev. Immunol. 2007, 7, 443–453.
- Lourenço, J.D.; Ito, J.T.; Martins MD, A.; Tibério ID FL, C.; Lopes, F.D.T.Q.D.S. Th17/Treg Imbalance in Chronic Obstructive Pulmonary Disease: Clinical and Experimental Evidence. Front. Immunol. 2021, 12, 804919.
- Zhang, L.; Cheng, Z.; Liu, W.; Wu, K. Expression of interleukin (IL)-10, IL-17A and IL-22 in serum and sputum of stable chronic obstructive pulmonary disease patients. COPD 2013, 10, 459–465.
- Sileikiene, V.; Laurinaviciene, A.; Lesciute-Krilaviciene, D.; Jurgauskiene, L.; Malickaite, R.; Laurinavicius, A. Levels of CD4+ CD25+ T Regulatory Cells in Bronchial Mucosa and Peripheral Blood of Chronic Obstructive Pulmonary Disease Indicate Involvement of Autoimmunity Mechanisms. Adv. Respir. Med. 2019, 87, 159–166.
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740.
- Yu, Z.X.; Ji, M.S.; Yan, J.; Cai, Y.; Liu, J.; Yang, H.F.; Zheng, J.X. The ratio of Th17/Treg cells as a risk indicator in early acute respiratory distress syndrome. Crit. Care 2015, 19, 82.
- Chen, L.; Yang, J.; Zhang, M.; Fu, D.; Luo, H.; Yang, X. SPP1 exacerbates ARDS via elevating Th17/Treg and M1/M2 ratios through suppression of ubiquitination-dependent HIF-1α degradation. Cytokine 2023, 164, 156107.
- Cheng, L.; Jiao, Y.; Jiang, W.; Zhang, X.; Zhang, L.; Jia, G. IL-33 Deficiency Attenuates Lung Inflammation by Inducing Th17 Response and Impacting the Th17/Treg Balance in LPS-Induced ARDS Mice via Dendritic Cells. J. Immunol. Res. 2022, 2022, 9543083.
- Urbankowski, T.; Hoser, G.; Domagała-Kulawik, J. Th1/Th2/Th17-related cytokines in the bronchoalveolar lavage fluid of patients with sarcoidosis: Association with smoking. Pol. Arch. Med. Wewn. 2012, 122, 320–325.
- Huang, H.; Lu, Z.; Jiang, C.; Liu, J.; Wang, Y.; Xu, Z. Imbalance between Th17 and Regulatory T-Cells in Sarcoidosis. Int. J. Mol. Sci. 2013, 14, 21463–21473.
- Mims, J.W. Asthma: Definitions and pathophysiology. Int. Forum Allergy Rhinol. 2015, 5, S2–S6.
- Chesné, J.; Braza, F.; Mahay, G.; Brouard, S.; Aronica, M.; Magnan, A. IL-17 in severe asthma. Where do we stand? Am. J. Respir. Crit. Care Med. 2014, 190, 1094–1101.
- Molet, S.; Hamid, Q.; Davoineb, F.; Nutku, E.; Tahaa, R.; Pagé, N.; Olivenstein, R.; Elias, J.; Chakir, J. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J. Allergy Clin. Immunol. 2001, 108, 430–438.
- Al-Ramli, W.; Préfontaine, D.; Chouiali, F.; Martin, J.G.; Olivenstein, R.; Lemière, C.; Hamid, Q. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J. Allergy Clin. Immunol. 2009, 123, 1185–1187.
- Barczyk, A.; Pierzchala, W.; Sozañska, E. Interleukin-17 in sputum correlates with airway hyperresponsiveness to methacholine. Respir. Med. 2003, 97, 726–733.
- Sun, Y.-C.; Zhou, Q.-T.; Yao, W.-Z. Sputum interleukin-17 is increased and associated with airway neutrophilia in patients with severe asthma. Chin. Med. J. 2005, 118, 953–956.
- Camargo, L.D.N.; dos Santos, T.M.; de Andrade, F.C.P.; Fukuzaki, S.; Lopes, F.D.T.Q.D.S.; Martins, M.D.A.; Prado, C.M.; Leick, E.A.; Righetti, R.F.; Tibério, I.D.F.L.C. Bronchial Vascular Remodeling Is Attenuated by Anti-IL-17 in Asthmatic Responses Exacerbated by LPS. Front. Pharmacol. 2020, 11, 1269.
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic Mucocutaneous Candidiasis in Humans with Inborn Errors of Interleukin-17 Immunity. Science 2011, 332, 65–68.
- Pandiyan, P.; Conti, H.R.; Zheng, L.; Peterson, A.C.; Mathern, D.R.; Hernández-Santos, N.; Edgerton, M.; Gaffen, S.L.; Lenardo, M.J. CD4+ CD25+ Foxp3+ Regulatory T Cells Promote Th17 Cells In Vitro and Enhance Host Resistance in Mouse Candida albicans Th17 Cell Infection Model. Immunity 2011, 34, 422–434.
- Rudner, X.L.; Happel, K.I.; Young, E.A.; Shellito, J.E. Interleukin-23 (IL-23)-IL-17 Cytokine Axis in Murine Pneumocystis carinii Infection. Infect. Immun. 2007, 75, 3055–3061.
- McKinley, L.; Logar, A.J.; McAllister, F.; Zheng, M.; Steele, C.; Kolls, J.K. Regulatory T Cells Dampen Pulmonary Inflammation and Lung Injury in an Animal Model of Pneumocystis Pneumonia. J. Immunol. 2006, 177, 6215–6226.
- Bai, H.; Gao, X.; Zhao, L.; Peng, Y.; Yang, J.; Qiao, S.; Zhao, H.; Wang, S.; Fan, Y.; Joyee, A.G.; et al. Respective IL-17A production by γδ T and Th17 cells and its implication in host defense against chlamydial lung infection. Cell Mol. Immunol. 2016, 14, 850–861.
- Thomas, R.; Wang, S.; Shekhar, S.; Peng, Y.; Qiao, S.; Zhang, C.; Shan, L.; Movassagh, H.; Gounni, A.S.; Yang, J.; et al. Semaphorin 3E Protects against Chlamydial Infection by Modulating Dendritic Cell Functions. J. Immunol. 2021, 206, 1251–1265.
- Thomas, R.; Wang, S.; Rashu, R.; Peng, Y.; Gounni, A.S.; Yang, X. Exogenous Semaphorin 3E treatment protects against chlamydial lung infection in mice. Front. Immunol. 2022, 13, 882412.
- Bai, H.; Cheng, J.; Gao, X.; Joyee, A.G.; Fan, Y.; Wang, S.; Yang, X. IL-17/Th17 promotes type 1 T cell immunity against pulmonary intracellular bacterial infection through modulating dendritic cell function. J. Immunol. 2009, 183, 5886–5895.
- Kelly, K.A.; Champion, C.I.; Jiang, J. The role of T regulatory cells in Chlamydia trachomatis genital infection. Chlamydia 2021, 91.
- Moore-Connors, J.M.; Fraser, R.; Halperin, S.A.; Wang, J. CD4+ CD25+ Foxp3+ Regulatory T Cells Promote Th17 Responses and Genital Tract Inflammation upon Intracellular Chlamydia muridarum Infection. J. Immunol. 2013, 191, 3430–3439.
- Marks, E.; Verolin, M.; Stensson, A.; Lycke, N. Differential CD28 and Inducible Costimulatory Molecule Signaling Requirements for Protective CD4+ T-Cell-Mediated Immunity against Genital Tract Chlamydia trachomatis Infection. Infect. Immun. 2007, 75, 4638–4647.
- Zhao, L.; Wang, H.; Thomas, R.; Gao, X.; Bai, H.; Shekhar, S.; Wang, S.; Yang, J.; Zhao, W.; Yang, X. NK cells modulate T cell responses via interaction with dendritic cells in Chlamydophila pneumoniae infection. Cell Immunol. 2020, 353, 104132.
- Li, J.; Dong, X.; Zhao, L.; Wang, X.; Wang, Y.; Yang, X.; Zhao, W. Natural killer cells regulate Th1/Treg and Th17/Treg balance in chlamydial lung infection. J. Cell Mol. Med. 2016, 20, 1339–1351.
This entry is offline, you can click here to edit this entry!