Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Substance Abuse
Alcohol abuse is a leading risk factor for the public health burden worldwide. Approved pharmacotherapies have demonstrated limited effectiveness over the last few decades in treating alcohol use disorders (AUD). A pathological downregulation of the metabotropic glutamate receptor 2 (mGlu2) in regulatory function has been observed in brain circuitries related to addictive behaviors and recent evidence suggest that pharmacological targeting of mGlu2 might yield therpeutic benefits for people with AUD.
- alcohol addiction
- metabotropic glutamate receptors
- serotonin 2a receptors
1. The Role of the Metabotropic Glutamate Receptor 2 in the Pathology of AUD
AUD, or substance use disorder (SUD) in general (DSM-5), is considered a brain disease defined by the repeated and continuous use of legal or illegal substances that cause clinically significant impairment [27]. For AUD, this means a systematically biased choice preference for alcohol at the expense of healthy rewards and continued use despite adverse consequences [28]. Neuropathologically, a model of altered neuroplasticity has been proposed as an underlying pathological mechanism [29]. Since glutamate is an important mediator of synaptic plasticity, it is no surprise that glutamate receptors are highly involved in the development and progression of substance dependence and alcoholism in particular [30,31,32].
Specifically, mGlu2 is highly abundant in the mesocorticolimbic and associated circuitries related to reward and drug seeking [33]. The prolonged exposure to drugs such as ethanol is believed to reduce mGlu2′s regulatory function in these systems, most notably in the medial prefrontal cortex (mPFC) and the nucleus accumbens (nAC), and by that contribute to the development of addiction-related behaviors [33]. For alcohol dependence in particular, a reduction of the GRM2 expression (the gene coding for mGlu2) was found in the anterior cingulate cortex (ACC) in patients with AUD as well as in the infralimbic cortex of rats with a history of alcohol dependence [34]. The role of mGlu2 in alcoholism was further strengthened by two studies in Grm2 mutant rats. First, it was demonstrated that the loss of functional mGlu2 in alcohol-preferring P rats due to the Grm2 cys407* mutation resulted in elevated alcohol consumption [35]. The second study discovered that the Grm2 cys407* mutation is also linked to excessive alcohol consumption in Hannover-derived Wistar rats [36]. Of note, one study did not find an association of prelimbic mGlu2 expression and high alcohol drinking [37]. Impaired mGlu2 function in the mPFC of rats resulted in excessive alcohol seeking and cognitive inflexibility, and these deficits were restored in postdependent rats—a well-established model of AUD [38]—by regional re-expression of the receptor [39].
2. The glutamatergic system in the brain.
Glutamate is considered the primary excitatory neurotransmitter of the central nervous system and has an important role in all aspects of brain function including the modulation of synaptic plasticity [40,41]. The release and concentration of glutamate are tightly regulated to ensure proper function at its targets, the glutamate receptors. These are divided into ionotropic glutamate receptors (iGlu) and metabotropic glutamate receptors (mGlu). While iGlu such as N-methyl-D-aspartate receptor (NMDAR), kainate and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid receptors (AMPAR) are ligand-gated ion channels that generate excitatory currents, mGlu are G-protein-coupled receptors (GPCRs) that activate second messenger signaling pathways and modulate synaptic transmission and plasticity on a longer scale [42]. The coordinated functions of iGlu and mGlu regulate the neural glutamate signaling, and dysfunctions or imbalances of these receptors, especially mGlu, have been implicated in various neuropsychiatric diseases [43,44].
mGlu exist as constitutive dimers and can be further subdivided into class I, II and III [45]. Class II consists of mGlu2 and mGlu3, which are distributed in various areas of the brain and are mostly detected at the synapse of neurons [43]. There, they are primarily localized in presynaptic locations (excluding the active zone [46]), where they inhibit the release of the neurotransmitter glutamate and gamma-aminobutyric acid (GABA) in excitatory glutamatergic neurons and inhibitory GABAergic interneurons, respectively [45]. Apart from its presynaptic localization, mGlu2/3 is also found throughout the axon, at the postsynaptic membrane and on glia cells [46,47,48,49,50]. The precise localization of mGlu at the synapse determines its synaptic functioning and is dependent on differential mechanisms of trafficking and positioning [51]. Furthermore, species differences have been observed for the localization of mGlu2 and mGlu3: in the primate PFC, mGlu2 is the predominant presynaptic receptor and mGlu3 the predominant postsynaptic receptor, while in the rat PFC both receptors are predominantly found on presynaptic terminals [50,52].
GPCRs mediate signaling through one of the four subfamilies of heteromeric G proteins (Gs, Gi/o, Gq/11 and G12/13), which consist of an α, β and γ subunit. mGlu2/3 canonically binds to Gi/o and activates related signaling pathways, depending on the molecular localization.
The activation of presynaptic and postsynaptic mGlu2/3 generally results in opposite effects (Figure 1). Downstream signaling pathways of presynaptic mGlu2/3 include the inhibition of adenylyl cyclase and the protein kinase A via Gi/o α subunit, as well as the inhibition of voltage-gated calcium channels (VGCCs) and the activation of G protein-coupled inward rectifying Potassium channels (GIRKs) via Gi/o βγ subunits [53,54,55]. Together, these signaling pathways inhibit the glutamate release in presynaptic glutamatergic neurons and thus provide a feedback mechanism to prevent excessive excitation and induce long-term depression (LTD). Activation of postsynaptic mGlu2/3 on the other site results in Gi/o-mediated NMDAR activation through various protein kinases including PKA, PKB, PKC and glycogen synthase kinase-3 beta (GSK-3B), as well as NMDAR trafficking by Snare proteins [56,57,58,59,60]. The activation of postsynaptic mGlu2/3 can further result in the increased surface expression of AMPAR via GSK-3B and extracellular signal-regulated kinases (ERKs) [61]. Taken together, postsynaptic mGlu2/3s potentiate the NMDAR current and regulate the NMDA/AMPA receptor trafficking, and thus cooperatively induce and maintain long-term potentiation (LTP). These findings demonstrate the sophisticated and opposing molecular mechanism of mGlu2/3 at presynaptic (LTD) and postsynaptic (LTP) locations required to ensure the correct and sustainable glutamatergic neurotransmission in the brain.
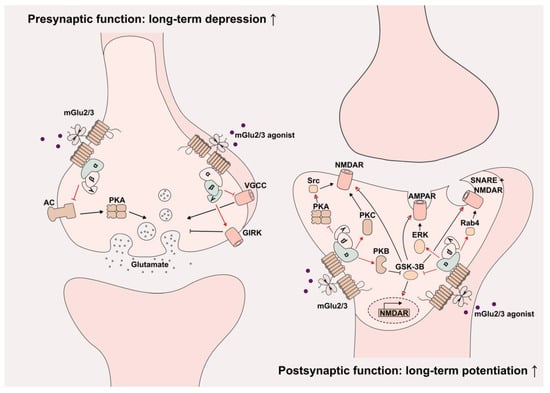
Figure 1. Molecular mechanisms of presynaptic and postsynaptic mGlu2/3 activation. Presynaptic (left) and postsynaptic (right) mGlu2 activation induces long-term depression and long-term potentiation, respectively. The relevant signaling cascades are displayed. Red indicates direct G-protein signaling consequences; red inhibitory arrow indicates second inhibition in the respective path. AC: Adenylyl cyclase, AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, ERK: Extracellular signal-regulated kinases, GIRK: G protein-coupled inward rectifying potassium channels, GSK-3B: Glycogen synthase kinase-3 beta, NMDAR: N-methyl-D-aspartate Receptor, PKA: Protein kinase A, PKB: Protein kinase B, PKC: Protein kinase C, Rab4: Ras-related protein Rab-4, Src: Proto-oncogene tyrosine–protein kinase Src and VGCC: Voltage-gated calcium channels.
3. mGlu2 as a therapeutic target
The virus-mediated rescue of mGlu2 in post-dependent rats was able to attenuate excessive alcohol seeking, suggesting the normalization of mGlu2 as a therapeutic opportunity [34]. AZD8529, a highly selective positive allosteric modulator (PAM) of mGlu2, was demonstrated to suppress cue-induced alcohol seeking responses in rats [62]. This effect was completely eliminated in rats lacking functional mGlu2, suggesting that the therapeutic effect is mediated by the activation of mGlu2 [62]. Furthermore, the administration of either the mGlu2 PAM LY487379 or one of the two mGlu2/3 agonists LY379268 and LY354740 to a well-established rat model of relapse demonstrated a significant decrease in relapse-like alcohol consumption [26]. This is in accordance with a prior study demonstrating reduced alcohol seeking and relapse behavior in alcohol-preferring rats after the administration of mGlu2 agonist LY404039 [63]. Craving and cognitive impairment can be rescued by restoring prefrontal mGlu2 levels [39]. In the same study, the administration of the psychedelic psilocybin was capable of restoring mGlu2 levels in alcohol-dependent rats. Given that psilocybin also reduced relapse behavior, the effect of the psychedelic on alcohol craving and drinking is, at least in part, mediated via mGlu2.
In sum, these studies demonstrate the pathological downregulation of mGlu2′s regulatory function in brain circuitries related to addictive behaviors and further pinpoint mGlu2 as a strong candidate for the treatment of AUD and preclinical trials targeting mGlu2 with agonists, and PAMs demonstrate promising results. Especially interesting in the light of the current clinical trials with psychedelics is the usage of psilocybin to target mGlu2 dysfunctions in alcohol-dependent rats. This molecular link between psilocybin and mGlu2 might at least partly explain the positive results observed in preclinical animal models and clinical studies for patients with AUD. The molecular mechanisms by which psilocybin and other psychedelics interact with or affect mGlu2 is only sparsely characterized, and further research is urgently needed for treatment development.
This entry is adapted from the peer-reviewed paper 10.3390/cells12060963
This entry is offline, you can click here to edit this entry!