Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
With an incidence of ten million cases and between one and two million deaths each year, Mycobacterium tuberculosis is the deadliest infectious agent currently. In this entry, the general physiopathology of tuberculosis is described, as well as the drugs constituting the first- and second-line treatments. The potential of nanosized drug delivery systems for the treatment of tuberculosis is also highlighted.
- mycobacterium tuberculosis
- tuberculosis
- granuloma
- antitubercular treatment
- antitubercular drugs
- antibioresistance
- drug delivery systems
- nanoparticles
- controlled release
- targeting
1. Tuberculosis Physiopathology
Rethinking and optimizing the treatment of TB requires a general understanding of its physiopathology. Before focusing on the behavior of the pathogen at the cellular level, it is necessary to comprehend how Mtb operates at the level of the entire organism. TB is transmitted by airborne droplets emitted by an individual suffering from an active form of the disease. During infection, the subject inhales bacilli, most of which are mechanically retained by mucus in the upper respiratory tract (usually with diameters higher than 5 µm) [1]. However, a small fraction (around 10%) reaches the lungs, bronchioles and alveoli, where alveolar macrophages then capture the bacteria [2]. At this stage, around 70% of subjects manage to eliminate the pathogen through the innate immune response. Otherwise, alveolar macrophages cross the lung epithelium to reach the interstitium [3].
In order to contain the infection, a characteristic cellular structure is then formed, typical of TB: the granuloma. The granuloma is a complex set of immune and inflammatory cells (such as macrophages, neutrophils, fibroblasts, T lymphocytes, B lymphocytes) that surrounds the infectious site and produces various cytokines and chemokines in order to maintain macrophage activation [4][5]. The purpose of the granuloma is not only to contain the proliferation of Mtb, but also to prevent its dissemination to other organs. What happens next is determined by the subject’s immunocompetence (Figure 1):
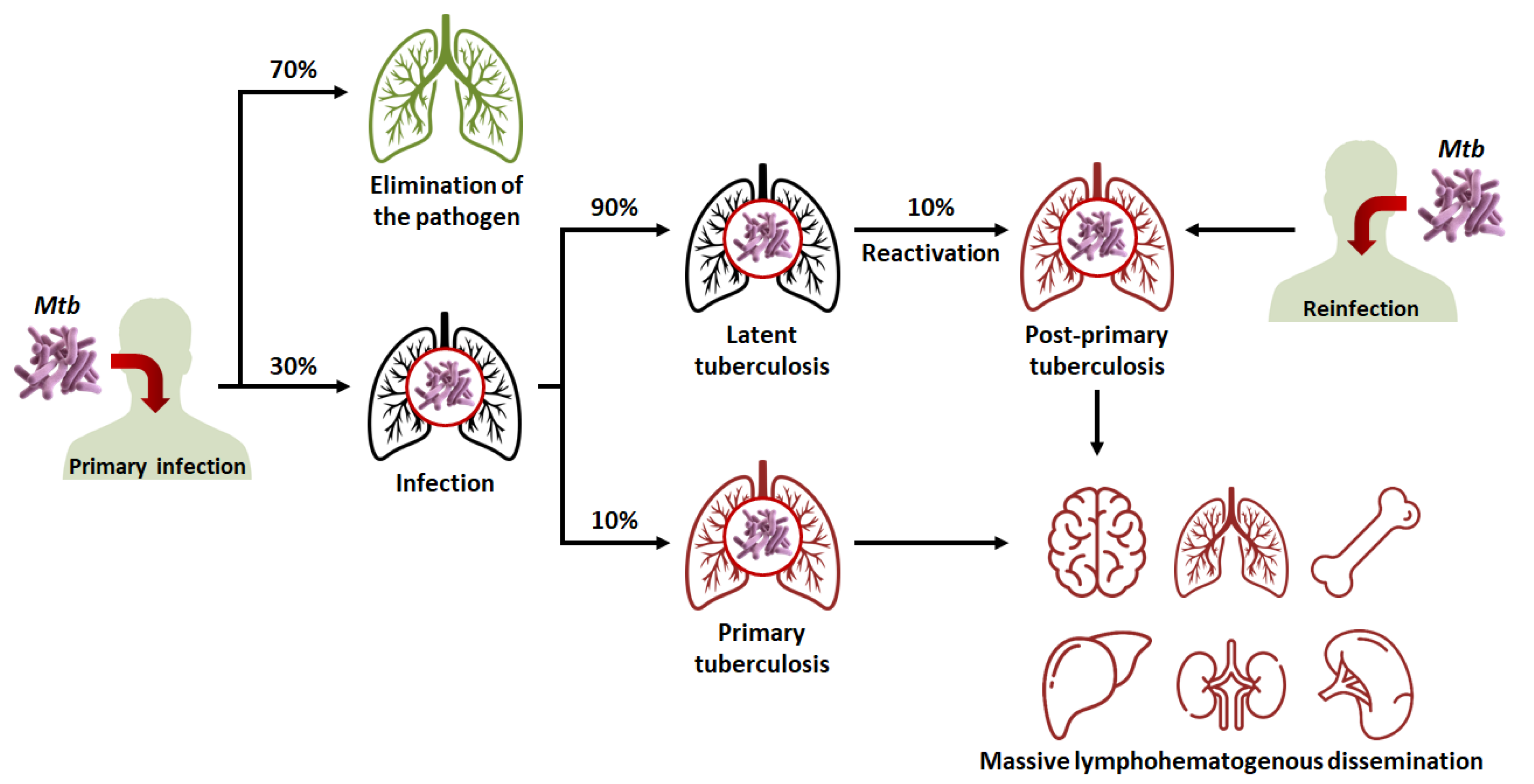
Figure 1. Evolution of the different clinical stages of tuberculosis (TB), from primary infection to miliary TB. Mtb: Mycobacterium tuberculosis.
-
In 90% of cases, the granuloma, acting as a physical and immunological barrier will succeed in stemming the infection. The pathogen will be contained in necrotic areas within granulomas located in the lungs [6]. Fibrous lesions will develop, and TB will evolve towards a latent form. This explains the fact that almost a third of the world’s population carries the pathogen, but that not more than ten million cases of active TB are diagnosed every year. Thus, most subjects that are latent cases (90%) will carry Mtb for decades, but will not show symptoms or infect other individuals [3].
-
In 10% of cases, most often in very young, elderly, or immunocompromised subjects, the granuloma will fail to contain Mtb. This phenomenon can also occur after the reactivation of dormant bacilli (which happens in 10% of latent cases) or with a novel inhalation of bacilli. The adaptive immune response will attempt to eliminate bacteria multiplying and escaping macrophages, but in doing so, will cause the destruction of lung tissue [7][8]. This will lead to the formation of caseous lesions and cavities, within which the growth of Mtb will no longer be controlled. TB will then evolve towards an active form. In certain cases, the pathogen will spread to other organs (brain, bones, liver, spleen, kidneys), and, in the most serious cases, the disease will evolve towards a miliary form, involving the massive lymphohematogenous dissemination of bacteria [9].
There is thus a balance between the latent form of TB, where the host’s immune system protects the subject while Mtb remains dormant within granulomas; and the active form, where Mtb proliferates to the detriment of its host. Thus, Koch’s bacillus is the “perfect” pathogen, in the sense that it preserves its host long enough for its own persistence while spreading through the population. In order to fully understand the success of Mtb’s “strategy”, it is necessary to study its behavior at the intracellular level. As previously said, after the inhalation of bacilli, the latter are phagocytosed by alveolar macrophages. Under physiological conditions, phagocytosis involves the acidification of the intracellular vesicle and its fusion with the lysosome (which allows its content to be degraded).
However, Mtb escapes the immune system by secreting various proteins which hinder the different stages of phagosomal maturation. Of note, it prevents the recruitment of GTPases and v-ATPases to the phagosomal membrane [4][5]. It also possesses a secretion system (ESX-1) that damages and permeabilizes the phagosomal membrane, which, in some cases, allows it to escape into the cytosol [10][11]. Thus, Mtb hijacks the defenses of its host to establish a niche inside of which it can either remain dormant or replicate, until causing the apoptosis or necrosis of the cell to disseminate germs and infect other cells [12].
2. Tuberculosis Treatment
Thanks to the effectiveness of the innate immune response, there have always been survivors of TB. However, the introduction of antibiotics to the market was a revolution which considerably reduced mortality rates over time [13]. As mentioned in the introduction, the discovery of the four first-line antitubercular antibiotics dates back more than half a century. Here presents their mechanisms of action (Figure 2):
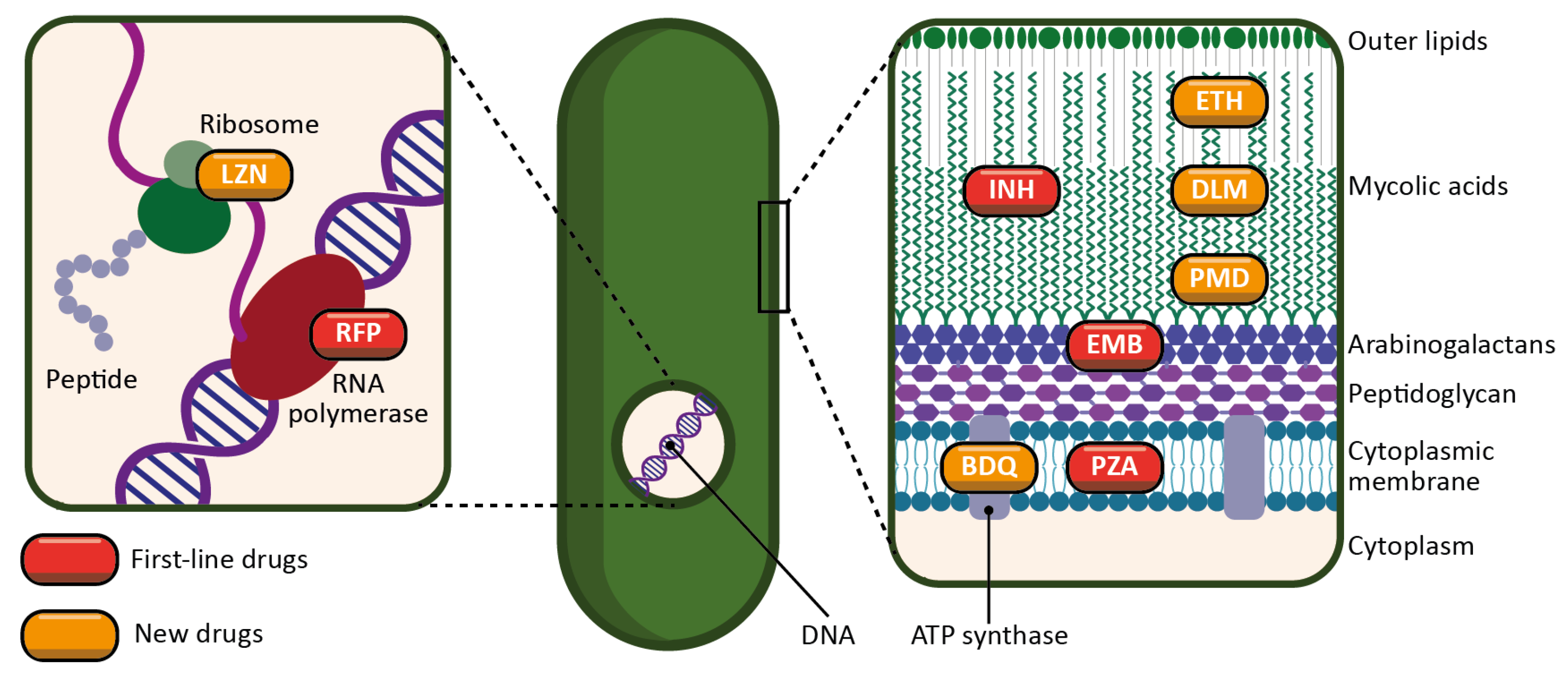
Figure 2. Schematic representation of the targets and mechanisms of action of the main antitubercular drugs. First-line drugs are represented in red (INH: isoniazid; RFP: rifampicin; PZA: pyrazinamide; EMB: ethambutol). New drugs, either newly discovered or repurposed, are represented in orange (ETH: ethionamide; LZN: linezolid; BDQ: bedaquiline; DLM: delamanid; PMD: pretomanid).
-
INH is a prodrug activated by a bacterial enzyme (KatG) [14]. The activation of the molecule produces an inhibitor of another bacterial enzyme, InhA, which results in the inhibition of mycolic acid synthesis, and therefore of the bacterial wall.
-
RFP is an inhibitor of the bacterial RNA polymerase, and thus acts by preventing protein synthesis [15]. It inhibits the elongation of bacterial RNA once it reaches two to three nucleotides in length.
-
PZA is a prodrug metabolized by a bacterial enzyme (pyrazinamidase) to become pyrazinoic acid [16]. The exact mechanism of action of pyrazinoic acid is still only partially elucidated, but the molecule is thought to act simultaneously on membrane energy production, the ribosomal protein RpsA, and other yet unidentified bacterial targets.
-
EMB targets arabinosyl transferase (a bacterial enzyme), thereby inhibiting arabinogalactan and bacterial wall synthesis [17]. EMB is also thought to exert a synergistic effect on INH activity.
The first-line treatment of TB is so effective that, in the case of a non-resistant strain and a therapy followed to completion, the risk of relapse is only 5%–8% [18][19]. Associated with INH, RFP reduces the duration of the therapy from eighteen to nine months. Taken for the first two months, PZA further reduces its duration by three months, leaving it at six months. EMB, finally, is used as an additional precaution in the event of unidentified resistance to one of the three main antibiotics. Thus, the treatment begins with a two-month induction phase involving the daily oral intake of the four antitubercular drugs. At the end of this phase, for most patients, no cultivable bacteria can be found in the sputum. The induction phase is followed by a four-month consolidation phase involving only INH and RFP to avoid possible relapse. Although there is considerable evidence of this treatment’s effectiveness, it can be prolonged and become more complex in the case of drug-resistant strains [20].
Drug resistance is a complex phenomenon involving an interplay of clinical, biological and microbiological processes [21]. In addition to the intrinsic resistance of bacteria, the lack of treatment adherence of patients also leads to the emergence of genetic resistance. Moreover, the complexity of granulomas is a barrier to the effective distribution of drugs, and therefore restrains their adequate supply. All this limits the use of first-line drugs and requires new antibiotics for therapy. There is a wide variety of drugs, usually used in combination, to treat cases of antibiotic-resistant TB. However, in the last twenty years, only four new drugs have been approved for this purpose: linezolid (LZN), bedaquiline (BDQ), delamanid (DLM) [21][22], and more recently, pretomanid (PMD) [23] (Figure 3). The mechanisms of action of some second-line drugs are described below (Figure 2):
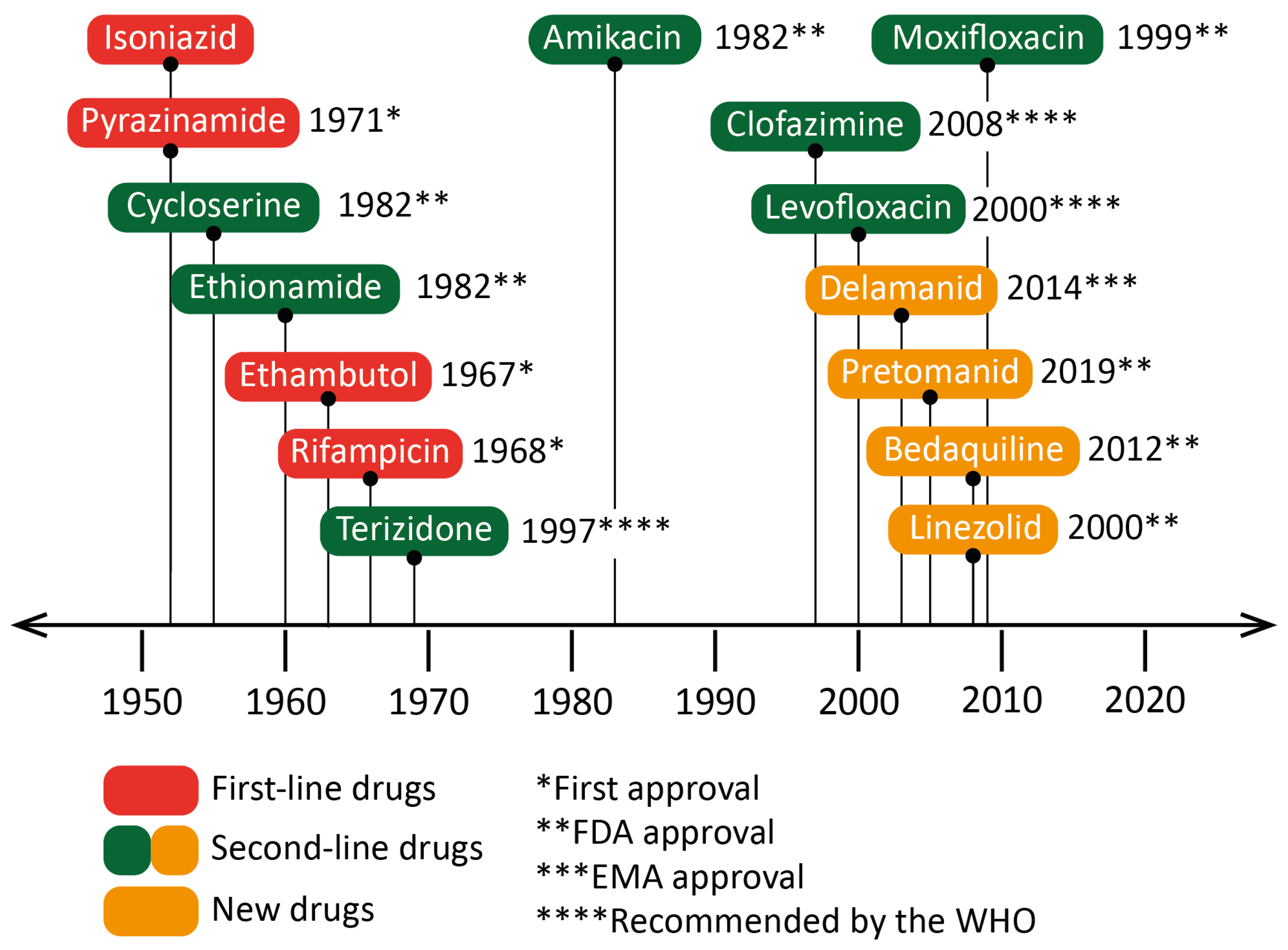
Figure 3. Dates of the first clinical trials for drugs recommended by the World Health Organization (WHO) for the treatment of tuberculosis (TB). The year of approval (or recommendation) for the use against Mycobacterium tuberculosis or against bacteria of the Mycobacterium genus is indicated next to the drug’s name. Isoniazid was accepted for prescription shortly after its first clinical trials. Some drugs, such as linezolid or moxifloxacin, were approved as broad-spectrum antibiotics, although specific clinical trials against TB occurred later. EMA: European Medicines Agency.
-
Ethionamide (ETH) is a thioisonicotinamide with a structure similar to that of INH [24]. ETH is a prodrug that, like INH, must be activated in order to inhibit mycobacterial fatty acid synthesis (by inhibiting enoyl-ACP reductase), which is essential for the production and repair of the bacterial cell wall.
-
LZN is a synthetic antimicrobial drug of the oxazolidinone class [25]. By binding to the rRNA on the 50S and 30S ribosomal subunits, it blocks the synthesis of bacterial proteins.
-
BDQ is the only FDA-approved antitubercular drug that targets the production of ATP [26]. BDQ inhibits the proton pumping mechanism by binding to the c subunit of the ATP synthase complex. It has also been observed that BDQ is able to act on the ε subunit of the enzyme.
-
DLM is a prodrug that, like INH, prevents the synthesis of mycolic acid in the bacterial cell wall [27]. DLM inhibits the synthesis of methoxy- and keto-mycolic acid by acting on the mycobacterial F420 system.
-
PMD is also a prodrug that acts under different mechanisms [28]. Under aerobic conditions, PMD inhibits protein and lipid synthesis by decreasing the availability of keto-mycolic acids through the inadequate oxidative transformation of the hydroxymycolate precursor. Under anaerobic conditions, PMD generates desnitro metabolites and provokes the release of nitric oxide, which inhibits cytochrome c oxidase and leads to a significant reduction in the amount of ATP present in bacteria.
In December 2022, the WHO released an update for the treatment of drug-resistant TB [29]. However, before addressing the new recommendations, it is necessary to define certain types of TB that are mentioned in the guidelines:
-
Multidrug-resistant TB (MDR-TB) is defined as TB with an RFP-resistant (RR-TB) and INH-resistant strain.
-
MDR/RR-TB stands for either MDR-TB or RR-TB.
-
Pre-extensively drug-resistant TB (pre-XDR-TB) is defined as MDR/RR-TB with resistance to at least one fluoroquinolone (either levofloxacin (LVX) or moxifloxacin (MOX)).
-
Extensively drug-resistant TB (XDR-TB) is defined as MDR/RR-TB with resistance to at least one fluoroquinolone (either LVX or MOX) and to at least one of the following two drugs: LZN and BDQ.
Thus, the updated recommendations are as follows:
-
For an INH-resistant only strain, the treatment is continued with RFP, PZA and EMB for a period of six months. INH is replaced by LVX.
-
For MDR/RR-TB and pre-XDR-TB:
-
Firstly, the WHO suggests adopting a six-month regimen (BPaLM) comprising LZN, BDQ, PMD and MOX (in the absence of a MOX-resistant strain). It is urged to use this new regimen instead of the nine-month or longer regimens for MDR/RR-TB, since BPaLM provides superior results in a shorter period.
-
Secondly, for MDR/RR-TB without a resistance to fluoroquinolones, the WHO recommends using a nine-month regimen rather than longer (eighteen-month) regimen. This regimen consists of BDQ (for six months) in combination with a fluoroquinolone (LVX or MOX), INH, PZA, EMB, ETH and clofazimine (CFZ) (for four months, with the possibility of extending this period to six months if the patient remains sputum smear-positive after four months), and then, a fluoroquinolone (LVX or MOX), PZA, EMB and CFZ (for five months). Two months of LZN might be used as an alternative to ETH.
-
-
For XDR-TB, the complementary molecules mentioned above constitute the core of the treatment.
Although the treatment of TB is theoretically effective, it is long, constraining and costly, even more in the case of antibiotic resistance (which concerns 20%–25% of clinical cases) [30]. However, it is precisely the complexity of the therapy that impedes patient compliance, and ultimately favors the emergence of resistant strains. In general, antitubercular drugs have low solubility, low metabolic stability, and low tissue penetration [31]. The challenge is to achieve therapeutic concentrations at the site of infection. The case of TB is troublesome, since multiple biological barriers stand in the way of active substances. Not only must the latter be directed towards the organ of interest (most often the lungs), but they must also cross the granulomas, the host cell membranes, and finally, the cell membranes of the pathogen hidden within infected cells [32].
Since the treatment is administered orally and diffuses systemically in the organism, high doses are required to reach sufficient concentrations at the site of infection. However, this therapy generates a strong accumulation of active molecules in non-targeted organs, and thus increases the side effects associated with these molecules. In 2019, Prasad et al. listed the side effects of antitubercular drugs, on the basis of more than a hundred articles and studies [33]. The first finding is that toxicity is more likely to occur during the induction phase rather than during the consolidation phase (which is expected, since the former involves more active molecules than the latter). The second finding is that first-line antibiotics present less toxicity (prevalence in 8%–85% of clinical cases) than second-line antibiotics (prevalence in 69%–96% of clinical cases). The third observation is that most of the side effects (gastrointestinal disorders, hepatotoxicity, peripheral neuropathy, optic neuritis, ototoxicity, nephrotoxicity, and skin reactions) are minor if they are managed at an early stage. However, poor patient monitoring can lead to irreversible damage, resulting in the interruption of the treatment, the selection of resistant strains, and finally, in a longer, more complex and more toxic treatment.
Based on the aforementioned observations, there is a clear challenge to optimize TB treatment. The hypothesis of the discovery of new active molecules which would surpass all pre-existing ones is unlikely. Both the scientific and medical communities are well aware that research has been going through a discovery void for the past decades. Indeed, the discovery of new molecules has considerably slowed down during this phase compared to the golden age of antibiotics. Optimizing the potential of current drugs would enable one to overcome many of the previously mentioned constraints. In this context, nanosized drug delivery systems (DDSs) have the potential to optimize the treatment’s efficiency while reducing its toxicity. Hundreds of publications illustrate the growing interest in this field. DDSs are used for the (co-)encapsulation of both first- and second-line drugs. DDSs could simultaneously (i) optimize the therapy’s antibacterial effects; (ii) reduce the doses; (iii) reduce the posology; (iv) diminish the toxicity; and as a global result, (v) mitigate the emergence of resistant strains.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics15020393
References
- Ryndak, M.B.; Laal, S. Mycobacterium Tuberculosis Primary Infection and Dissemination: A Critical Role for Alveolar Epithelial Cells. Front. Cell. Infect. Microbiol. 2019, 9, 299.
- Philips, J.A.; Ernst, J.D. Tuberculosis Pathogenesis and Immunity. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 353–384.
- Lin, P.L.; Flynn, J.L. Understanding Latent Tuberculosis: A Moving Target. J. Immunol. 2010, 185, 15–22.
- Guirado, E.; Schlesinger, L.S.; Kaplan, G. Macrophages in Tuberculosis: Friend or Foe. Semin. Immunopathol. 2013, 35, 563–583.
- Kinsella, R.L.; Zhu, D.X.; Harrison, G.A.; Mayer Bridwell, A.E.; Prusa, J.; Chavez, S.M.; Stallings, C.L. Perspectives and Advances in the Understanding of Tuberculosis. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 377–408.
- Hunter, R.L. Pathology of Post Primary Tuberculosis of the Lung: An Illustrated Critical Review. Tuberculosis 2011, 91, 497–509.
- Grotz, E.; Tateosian, N.; Amiano, N.; Cagel, M.; Bernabeu, E.; Chiappetta, D.A.; Moretton, M.A. Nanotechnology in Tuberculosis: State of the Art and the Challenges Ahead. Pharm. Res. 2018, 35, 213.
- Hunter, R.L. The Pathogenesis of Tuberculosis: The Early Infiltrate of Post-Primary (Adult Pulmonary) Tuberculosis: A Distinct Disease Entity. Front. Immunol. 2018, 9, 2108.
- Sharma, S.K.; Mohan, A. Miliary Tuberculosis. In Tuberculosis and Nontuberculous Mycobacterial Infections; Schlossberg, D., Ed.; ASM Press: Washington, DC, USA, 2017; pp. 491–513.
- Blischak, J.D.; Tailleux, L.; Mitrano, A.; Barreiro, L.B.; Gilad, Y. Mycobacterial Infection Induces a Specific Human Innate Immune Response. Sci. Rep. 2015, 5, 16882.
- Simeone, R.; Sayes, F.; Song, O.; Gröschel, M.I.; Brodin, P.; Brosch, R.; Majlessi, L. Cytosolic Access of Mycobacterium Tuberculosis: Critical Impact of Phagosomal Acidification Control and Demonstration of Occurrence In Vivo. PLoS Pathog. 2015, 11, e1004650.
- Queval, C.J.; Brosch, R.; Simeone, R. The Macrophage: A Disputed Fortress in the Battle against Mycobacterium Tuberculosis. Front. Microbiol. 2017, 8, 2284.
- Gould, K. Antibiotics: From Prehistory to the Present Day. J. Antimicrob. Chemother. 2016, 71, 572–575.
- Lei, B.; Wei, C.-J.; Tu, S.-C. Action Mechanism of Antitubercular Isoniazid. J. Biol. Chem. 2000, 275, 2520–2526.
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. Cell 2001, 104, 901–912.
- Zhang, Y.; Shi, W.; Zhang, W.; Mitchison, D. Mechanisms of Pyrazinamide Action and Resistance. Microbiol. Spectr. 2014, 2, 2.
- Zhu, C.; Liu, Y.; Hu, L.; Yang, M.; He, Z.-G. Molecular Mechanism of the Synergistic Activity of Ethambutol and Isoniazid against Mycobacterium Tuberculosis. J. Biol. Chem. 2018, 293, 16741–16750.
- Horsburgh, C.R.; Barry, C.E.; Lange, C. Treatment of Tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160.
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis. Module 4: Treatment—Drug-Susceptible Tuberculosis Treatment; World Health Organization: Geneva, Switzerland, 2022.
- Bahuguna, A.; Rawat, D.S. An Overview of New Antitubercular Drugs, Drug Candidates, and Their Targets. Med. Res. Rev. 2020, 40, 263–292.
- Singh, V.; Chibale, K. Strategies to Combat Multi-Drug Resistance in Tuberculosis. Acc. Chem. Res. 2021, 54, 2361–2376.
- Olaru, I.D.; von Groote-Bidlingmaier, F.; Heyckendorf, J.; Yew, W.W.; Lange, C.; Chang, K.C. Novel Drugs against Tuberculosis: A Clinician’s Perspective. Eur. Respir. J. 2015, 45, 1119–1131.
- Provisional CDC Guidance for the Use of Pretomanid as Part of a Regimen to Treat Drug-Resistant Tuberculosis Disease. Available online: https://www.cdc.gov/tb/topic/drtb/bpal/default.htm (accessed on 18 November 2022).
- Vale, N.; Gomes, P.; Santos, H.A. Metabolism of the Antituberculosis Drug Ethionamide. Curr. Drug Metab. 2013, 14, 151–158.
- Oehadian, A.; Santoso, P.; Menzies, D.; Ruslami, R. Concise Clinical Review of Hematologic Toxicity of Linezolid in Multidrug-Resistant and Extensively Drug-Resistant Tuberculosis: Role of Mitochondria. Tuberc. Respir. Dis. 2022, 85, 111–121.
- Khoshnood, S.; Goudarzi, M.; Taki, E.; Darbandi, A.; Kouhsari, E.; Heidary, M.; Motahar, M.; Moradi, M.; Bazyar, H. Bedaquiline: Current Status and Future Perspectives. J. Glob. Antimicrob. Resist. 2021, 25, 48–59.
- Khoshnood, S.; Taki, E.; Sadeghifard, N.; Kaviar, V.H.; Haddadi, M.H.; Farshadzadeh, Z.; Kouhsari, E.; Goudarzi, M.; Heidary, M. Mechanism of Action, Resistance, Synergism, and Clinical Implications of Delamanid Against Multidrug-Resistant Mycobacterium Tuberculosis. Front. Microbiol. 2021, 12, 717045.
- Occhineri, S.; Matucci, T.; Rindi, L.; Tiseo, G.; Falcone, M.; Riccardi, N.; Besozzi, G. Pretomanid for Tuberculosis Treatment: An Update for Clinical Purposes. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100128.
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis. Module 4: Treatment—Drug-Resistant Tuberculosis Treatment, 2022 Update; World Health Organization: Geneva, Switzerland, 2022.
- Dean, A.S.; Cox, H.; Zignol, M. Epidemiology of Drug-Resistant Tuberculosis. In Strain Variation in the Mycobacterium tuberculosis Complex: Its Role in Biology, Epidemiology and Control; Gagneux, S., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 1019, pp. 209–220.
- Sarkar, K.; Kumar, M.; Jha, A.; Bharti, K.; Das, M.; Mishra, B. Nanocarriers for Tuberculosis Therapy: Design of Safe and Effective Drug Delivery Strategies to Overcome the Therapeutic Challenges. J. Drug Deliv. Sci. Technol. 2022, 67, 102850.
- Tanner, L.; Denti, P.; Wiesner, L.; Warner, D.F. Drug Permeation and Metabolism in Mycobacterium Tuberculosis: Prioritising Local Exposure as Essential Criterion in New TB Drug Development. IUBMB Life 2018, 70, 926–937.
- Prasad, R.; Singh, A.; Gupta, N. Adverse Drug Reactions in Tuberculosis and Management. Indian J. Tuberc. 2019, 66, 520–532.
This entry is offline, you can click here to edit this entry!