Epilepsy is a common brain disease characterized as a long-lasting propensity to engender epileptic seizures. These are defined by the International League Against Epilepsy (ILAE), as a transient occurrence of signs and/or symptoms, due to abnormal excessive or synchronous neuronal activity in the brain. The ILAE also describes that patients with epilepsy, besides neurobiological problems, also face cognitive, psychological, and social issues.
Epileptic seizures can be classified depending on the onset (focal, generalized or unknown) and depending on the etiology (genetic, structural, infectious, metabolic, immune, unknown, or with more than one etiology).
Epileptogenesis is when a physiological and functional brain develops recurrent and unprovoked seizures, due to abnormal biological alterations. Epileptogenesis encompasses: The moment a precipitating injury (such as stroke or traumatic brain injury) or event (as status epilepticus (SE) or febrile seizure) occurs; the latent period between this epileptogenic insult and a modified epileptic brain (having spontaneous unprovoked seizures); and the mechanisms that occur during chronic epilepsy.
Despite that the knowledge about the epileptogenesis process has significantly increased, most of the current drugs for epilepsy are used to treat symptoms, meaning to stop the seizures. These drugs, named AEDs, do not prevent or cure epilepsy. Therefore, finding drugs that work as antiepileptogenic, interreacting with the process of epilepsy development, is fundamental.
- Epilepsy
- Mesial temporal lobe epilepsy
- Seizures
- Antiepileptic drugs
- Antiepileptogenic drugs
- Epileptogenesis
- Experimental models
- Neurological disease
1. Introduction
Epilepsy, one of the oldest known neurological diseases[1], was, for centuries, associated with a divine malady or demonic possession—and as such, exorcism was the only known therapy for this pathology[2]. Hippocrates in 400 B.C. demystified epilepsy by arguing that it was a medical problem that originated in the brain, instead of a problem of divine origin[1]. However, the prevailing supernatural view remained rather unchanged until the 17th century, when the first effective antiepileptic medicine, bromide, was introduced[3]. Antiepileptic drugs (AEDs), focal epilepsy surgery, vagus nerve stimulation, and the ketogenic diet are the available therapies to treat epilepsy. Nevertheless, the effectiveness of these therapies is highly affected by disease etiology (reviewed in Reference[4]).
2. Epilepsy and Epileptogenesis
Epilepsy is a common brain disease, affecting between 50 to 65 million people worldwide[5][6][7][8], and is characterized as a long-lasting propensity to engender epileptic seizures. These are defined by the International League Against Epilepsy (ILAE), as a transient occurrence of signs and/or symptoms, due to abnormal excessive or synchronous neuronal activity in the brain[9]. The ILAE also describes that patients with epilepsy, besides neurobiological problems, also face cognitive, psychological, and social issues[9].
Epileptic seizures can be classified depending on the onset as: Focal, if limited to one hemisphere; generalized, if rapidly spread bilaterally; or unknown, if the onset is unable to be determined, due to lack of information[10][11]. Under this big umbrella, seizures can also be categorized as motor or nonmotor, and they can be detailed depending on awareness (in case of focal seizures)[11]. In a broad view, motor behavior may include loss of tone (atonic), sustained stiffening (tonic), rhythmic jerks (clonic), irregular and brief jerks (myoclonic), flexion, or extension of arms, and flexion of trunk (epileptic spasms). Generalized motor seizures can comprise more than one motor behavior. Generalized non-motor seizures imply absence seizures. The type of epilepsy can also be classified as focal, generalized, combined generalized and focal, or unknown. Besides seizure and epilepsy type, diagnosis also comprises the recognition of epileptic syndromes (such as childhood absence epilepsy, Lennox-Gastaut syndrome, or Dravet syndrome)[10].
It is also important to know the etiology of epilepsy to adequate the best treatment. Epilepsy can be classified with different etiologies: Genetic, structural, infectious, metabolic, immune, unknown, or with more than one etiology[10]. Genetic etiology refers to seizures caused by a genetic mutation, such as in Dravet syndrome. Structural etiology implies the presence of acquired or abnormal genetic structures, such as in the hippocampus or amygdala, often associated with mesial Temporal Lobe Epilepsy (mTLE), which is the most common and studied form of epilepsy, and is frequently intractable. An infectious etiology is related to an infection, like cerebral malaria or the Zika virus, resulting in the appearance of seizures. A metabolic etiology is linked to metabolic disorders, such as porphyria. Finally, an immune origin is usually associated with central nervous system (CNS) inflammation mediated by autoimmune disorders (for example, autoimmune encephalitis)[10].
Epileptogenesis is when a physiological and functional brain develops recurrent and unprovoked seizures, due to abnormal biological alterations (reviewed in References[12][13]). Epileptogenesis encompasses: The moment a precipitating injury (such as stroke or traumatic brain injury) or event (as status epilepticus (SE) or febrile seizure) occurs; the latent period between this epileptogenic insult and a modified epileptic brain (having spontaneous unprovoked seizures); and the mechanisms that occur during chronic epilepsy (reviewed in References[12][13]) (Figure 1). It is worth mention that traditionally, the process of epileptogenesis was considered to stop at the time of the first spontaneous seizure (reviewed in Reference[14]).
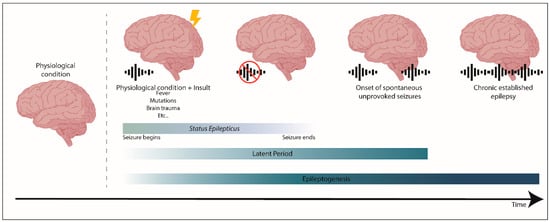
Using mainly experimental models of mTLE it was possible to start unveiling the typical alterations that are involved in epileptogenesis[12][14][15]. However, the mechanisms underlying epileptogenesis are not yet fully understood. It is still not known which mechanisms are responsible for the generation of epilepsy or which are secondary or compensatory mechanisms that intend to repair the brain. The most frequent alterations observed in experimental mTLE models are neuronal cell death, mainly of hippocampal pyramidal cells[16], but also of hilar mossy cells, which are excitatory neurons in the hilus of the dentate gyrus[16], and of inhibitory GABAergic interneurons[17]; reactive gliosis[18]; blood–brain barrier damage[19]; alterations in the expression of GABAA receptor subunit[20]; and changes in diverse signaling pathways, for example, brain-derived neurotrophic factor/tyrosine receptor kinase B (BDNF/TrkB), mammalian target of rapamycin (mTOR), or Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathways[21] (reviewed in References[13][22][23][24][25]). Another important feature is the modifications in neurogenesis and associated-processes[16], which will be further detailed in this entry.
Despite that the knowledge about the epileptogenesis process has significantly increased, most of the current drugs for epilepsy are used to treat symptoms, meaning to stop the seizures. These drugs, named AEDs, do not prevent or cure epilepsy (reviewed in Reference[26]). Therefore, finding drugs that work as antiepileptogenic, interreacting with the process of epilepsy development, is fundamental. Current AEDs are mainly based on four mechanisms of action: (1) Modulation of voltage-gated ion channels (as valproic acid, phenytoin or carbamazepine); (2) enhancement of GABA-mediated inhibitory neurotransmission (like valproic acid, phenobarbital or tiagabine); (3) reduction of glutamate-mediated excitatory neurotransmission (as felbamate, perampanel or gabapentin) and (4) modulation of neurotransmitter release through presynaptic release machinery (as levetiracetam, gabapentin or pregabalin) (reviewed in References[24][26]).
Given the fact that about 30% of patients with epilepsy remain resistant to pharmacotherapy, continuing to experience seizures (reviewed in References[27][28]), it is imperative to persist studying the mechanisms underlying epileptogenesis. Developing innovative antiepileptogenic therapies that can modify this process, instead of only diminishing or abolishing seizures, and can decrease epilepsy-related comorbidities after the clinical diagnosis of epilepsy, is critical.
Experimental models, either in vivo or in vitro, mimic different types of epileptic seizures, syndromes, or specific aspects of the disease. In vivo animal models have been categorized into a different seizure or epilepsy models: Chemical or pharmacological (induced by pilocarpine, kainate or pentylenetetrazole (PTZ)); electrical stimulation (such as the kindling model or maximal electroshock seizures (MES)); genetic (mutations related to dysfunction of ion channels, receptors, enzymes or transporters); developmental (like the febrile seizures model) and trauma (as cortical undercut model) (reviewed in References[29][30][31]). These models are classified as models of epilepsy or models of seizures, depending on whether they result in chronic epilepsy or not, respectively[29].
The most commonly used models to mimic mTLE, are the pilocarpine (an acetylcholine receptor agonist), kainate (a glutamate analogue), or kindling models (a process that triggers epileptic seizures through repeated low-intensity electrical stimulation in a given brain region) (reviewed in References[30][32]). Pilocarpine- and kainate-induced SE models are more similar with the epileptic process occurring in humans than the kindling model, since they include an initial precipitating injury, a latent period, and finally, spontaneous, recurrent chronic seizures (reviewed in Reference[29]). On the other hand, the kindling model enhances seizure susceptibility, potentiating the generalization of electrical-induced seizures to other areas of the brain and ultimately, promoting spontaneous seizures (reviewed in Reference[30]).
In vitro models are mainly models of epileptogenesis, such as organotypic brain slices, or models of epileptiform activity, in which the biological preparations, like neuronal cultures or acute brain slices, are susceptible to chemoconvulsants, and therefore, epileptiform activity is acutely induced (reviewed in Reference[33]).
This entry is adapted from the peer-reviewed paper 10.3390/ijms21197309
References
- Emmanouil Magiorkinis; Kalliopi Sidiropoulou; Aristidis Diamantis; Hallmarks in the history of epilepsy: Epilepsy in antiquity. Epilepsy & Behavior 2010, 17, 103-108, 10.1016/j.yebeh.2009.10.023.
- World Health Organization. WHO|Atlas: Epilepsy Care in the World; Global Campaign against Epilepsy, International Bureau of Epilepsy, International League against Epilepsy, Eds.; WHO: Geneva, Switzerland, 2005; ISBN 978-92-4-156303-1.
- Piero Perucca; Frank G Gilliam; Adverse effects of antiepileptic drugs. The Lancet Neurology 2012, 11, 792-802, 10.1016/s1474-4422(12)70153-9.
- Mark Manford; Recent advances in epilepsy. Journal of Neurology 2017, 264, 1811-1824, 10.1007/s00415-017-8394-2.
- WHO. Epilepsy: A Public Health Imperative; WHO: Geneva, Switzerland, 2019; ISBN 978-92-4-151593-1.
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the burden of active and life-time epilepsy: A meta-analytic approach. Epilepsia 2010, 51, 883–890.
- Fiest, K.M.; Sauro, K.M.; Wiebe, S.; Patten, S.B.; Kwon, C.-S.; Dykeman, J.; Pringsheim, T.; Lorenzetti, D.L.; Jetté, N. Prevalence and incidence of epilepsy: A systematic review and meta-analysis of international studies. Neurology 2017, 88, 296–303.
- Feigin, V.L.; Abajobir, A.A.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abera, S.F.; Abyu, G.Y.; Ahmed, M.B.; Aichour, A.N.; Aichour, I.; et al. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897.
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J. Epileptic Seizures and Epilepsy: Definitions Proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472.
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521.
- Fisher, R.S.; Cross, J.H.; D’Souza, C.; French, J.A.; Haut, S.R.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia 2017, 58, 531–542.
- Pitkänen, A.; Lukasiuk, K.; Dudek, F.E.; Staley, K.J. Epileptogenesis. Cold Spring Harb. Perspect. Med. 2015, 5, a022822.
- Goldberg, E.M.; Coulter, D.A. Mechanisms of epileptogenesis: A convergence on neural circuit dysfunction. Nat. Rev. Neurosci. 2013, 14, 337–349.
- Asla Pitkänen; Katarzyna Lukasiuk; Mechanisms of epileptogenesis and potential treatment targets. The Lancet Neurology 2011, 10, 173-186, 10.1016/s1474-4422(10)70310-0.
- Pavel Klein; Raymond Dingledine; Eleonora Aronica; Christophe Bernard; Ingmar Blümcke; Detlev Boison; Martin J. Brodie; Amy R. Brooks-Kayal; Jerome Engel; Patrick A. Forcelli; et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate?. Epilepsia 2017, 59, 37-66, 10.1111/epi.13965.
- Rao, M.S.; Hattiangady, B.; Reddy, D.S.; Shetty, A.K. Hippocampal neurodegeneration, spontaneous seizures, and mossy fiber sprouting in the F344 rat model of temporal lobe epilepsy. J. Neurosci. Res. 2006, 83, 1088–1105.
- Kobayashi, M.; Buckmaster, P.S. Reduced Inhibition of Dentate Granule Cells in a Model of Temporal Lobe Epilepsy. J. Neurosci. 2003, 23, 2440–2452.
- Muro-García, T.; Martín-Suárez, S.; Espinosa, N.; Valcárcel-Martín, R.; Marinas, A.; Zaldumbide, L.; Galbarriatu, L.; Sierra, A.; Fuentealba, P.; Encinas, J.M. Reactive Disruption of the Hippocampal Neurogenic Niche After Induction of Seizures by Injection of Kainic Acid in the Amygdala. Front. Cell Dev. Biol. 2019, 7, 158.
- van Vliet, E.A.; da Costa Araujo, S.; Redeker, S.; van Schaik, R.; Aronica, E.; Gorter, J.A. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 2007, 130, 521–534.
- Brooks-Kayal, A.R.; Shumate, M.D.; Jin, H.; Rikhter, T.Y.; Coulter, D.A. Selective changes in single cell GABA A receptor subunit expression and function in temporal lobe epilepsy. Nat. Med. 1998, 4, 1166–1172.
- Laura Ester Montroull; Víctor Danelon; Andrea Beatriz Cragnolini; Daniel Mascó; Loss of TrkB Signaling Due to Status Epilepticus Induces a proBDNF-Dependent Cell Death. Frontiers in Cellular Neuroscience 2019, 13, 4, 10.3389/fncel.2019.00004.
- Lévesque, M.; Avoli, M. The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav. Rev. 2013, 37, 2887–2899.
- Scharfman, H.E.; Myers, C.E. Hilar mossy cells of the dentate gyrus: A historical perspective. Front. Neural Circuits 2013, 6.
- Reddy, D.S.; Kuruba, R. Experimental models of status epilepticus and neuronal injury for evaluation of therapeutic interventions. Int. J. Mol. Sci. 2013, 14, 18284–18318.
- Swissa, E.; Serlin, Y.; Vazana, U.; Prager, O.; Friedman, A. Blood–brain barrier dysfunction in status epileptics: Mechanisms and role in epileptogenesis. Epilepsy Behav. 2019, 101, 106285.
- Graeme J. Sills; Michael A. Rogawski; Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966, 10.1016/j.neuropharm.2020.107966.
- Clossen, B.L.; Reddy, D.S. Novel therapeutic approaches for disease-modification of epileptogenesis for curing epilepsy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1519–1538.
- Billakota, S.; Devinsky, O.; Kim, K.-W. Why we urgently need improved epilepsy therapies for adult patients. Neuropharmacology 2019, 18, 107855.
- Kandratavicius, L.; Alves Balista, P.; Lopes-Aguiar, C.; Ruggiero, R.N.; Umeoka, H.; Garcia-Cairasco, N.; Soares Bueno-Junior, L.; Pereira Leite, J. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705.
- Raol, Y.H.; Brooks-Kayal, A.R. Experimental Models of Seizures and Epilepsies. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2012; Volume 105, pp. 57–82. ISBN 978-0-12-394596-9.
- Löshcer, W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 2002, 50, 105–123.
- Matthew R. Sarkisian; Overview of the Current Animal Models for Human Seizure and Epileptic Disorders. Epilepsy & Behavior 2001, 2, 201-216, 10.1006/ebeh.2001.0193.
- Massimo Avoli; John G.R. Jefferys; Models of drug-induced epileptiform synchronization in vitro. Journal of Neuroscience Methods 2016, 260, 26-32, 10.1016/j.jneumeth.2015.10.006.