1. Introduction
Chemotherapy is mainly reserved for cases not eligible for curative therapy for hepatocellular carcinoma such as resection, transplantation, or ablation, and so serves mainly as a palliative measure [
20]. Numerous chemotherapeutic drugs have been assessed in the treatment of HCC. Few are consistently linked to antitumor responses [
21]. Chemotherapy can be given systemically or locally. Regional chemotherapy may also comprise intra-arterial treatment, which has a similar effect to chemoembolization. HCC is typically linked with cirrhosis, which limits the dose and response rate of systemic chemotherapy (generally fewer than 25% of objective responses) [
22]. Antiangiogenic medicines have significant potential for treating HCC, owing to the tumor’s vascularity [
23].
Chemotherapy has not been frequently utilized in patients with advanced HCC since HCC has generally been regarded as a chemotherapy-resistant malignancy, and systemic chemotherapy is typically not tolerated well in patients with significant underlying liver disease [
24]. Chemotherapy might still be indicated in some patients, especially those with an underlying non-cirrhotic liver. The Eastern Cooperative Oncology Group Performance Status Scale recommends systemic treatment for progressed stage (C) HCC with portal hypertension, extrahepatic dissemination, and intact liver activity [
24]. Cases involving advanced-stage (C) patients with HCC who are ineligible for TACE therapy may be a better match for this approach. For the first time, targeted therapy and immunotherapy have emerged as therapeutic options for all stages of the disease as shown in
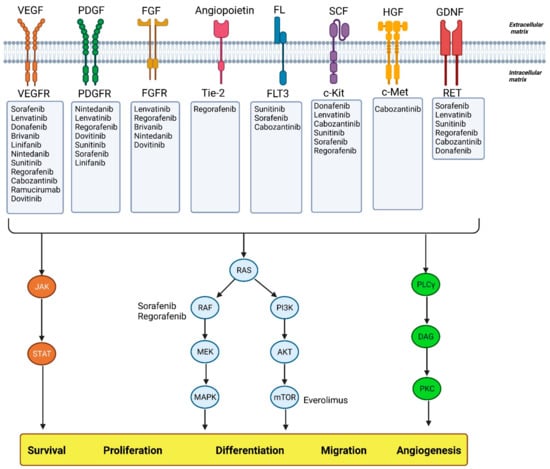
Figure 1 [
25]. Current FDA-approved agents for targeted therapy in advanced HCC are summarized in
Table 1.
Figure 1. Agents used in targeted therapy of HCC and their different pathways. Abbreviations: VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor; PDGF: platelet-derived growth factor; PDGFR: platelet-derived growth factor receptor; FGF: fibroblast growth factor; FGFR: fibroblast growth factor receptors; Tie-2: an angiopoietin receptor; FL: Fms-like tyrosine kinase 3 ligand; FLT3: Fms-like tyrosine kinase 3; SCF: stem cell factor; HGF: hepatocyte growth factor; c-Met: mesenchymal-epithelial transition factor; GDNF: glial cell-derived neurotrophic factor; JAK: Janus kinases; STAT: signal transducer and activator of transcription proteins; Ras: rat sarcoma virus; Raf: rapidly accelerated fibrosarcoma; MEK: mitogen-activated protein kinase; MAPK: mitogen activated protein kinases; PI3K: phosphoinositide 3-kinases; AKT: protein kinase B; mTOR: mammalian target of rapamycin; PLCγ: phospholipase C γ; DAG: diacylglycerol; PKC: protein kinase C.
Table 1. Selected first-line and second-line FDA-approved agents for targeted therapies in advanced HCC.
2. First Line
2.1. Sorafenib
Sorafenib, an orally administered multikinase inhibitor, is the first systemic drug demonstrated to prolong survival in advanced HCC [
37,
38]. It delays disease progression by influencing two key signaling pathways. By inhibiting molecular components of the Raf, MEK, and ERK signaling pathways, it leads to diminished tumor growth, and by suppressing VEGFR-1, VEGFR-2, VEGFR-3, and PDGFR-β, it stops neovascularization [
39]. Sorafenib monotherapy extended overall survival (OS) and delayed the time to progression (TTP) in patients with HCC, according to the results of two well-designed, randomized, double-blind, placebo-controlled, and multinational phase III trials. The findings of the SHARP study, which was conducted in Europe, America, Australia, and New Zealand and included a few Asian patients, were subsequently confirmed by the Asia-Pacific study results. Both trials showed no significant difference between sorafenib and placebo in the time to symptomatic progression (TTSP). The disease control rate was significantly higher with sorafenib versus placebo in both studies. No complete responses were seen in either trial, and partial response rates were very low. Overall, both studies indicated that sorafenib was effective in prolonging 3 months of median overall survival in patients with late-stage HCC [
37,
38]. GIDEON was a large, prospective, observational study conducted in 39 countries among 2708 HCC patients that revealed regional variations in the management of HCC and in patient outcomes. The study showed that the safety profile of sorafenib was consistent across patients with preserved liver function and those in whom the liver was not functioning properly, suggesting that sorafenib may be a valid treatment for some patients with liver impairment [
40,
41,
42]. The STELLA and SOFIA studies conducted in Italy and the INSIGHT studies conducted in Germany and Austria were prospective, multicenter, and non-interventional studies that demonstrated the efficacy of sorafenib in HCC patients in real-world settings [
43,
44,
45]. Sorafenib was generally well tolerated in patients with aHCC, with a manageable adverse effect profile. The most common sorafenib treatment-related adverse events were diarrhea, hand-foot skin reactions, hypertension, anorexia, alopecia, weight loss, dry skin, and abdominal pain [
38]. However, drug resistance limits the therapeutic effect of sorafenib, so that only about 30% of HCC patients acquired benefits from sorafenib and the development of resistance within 6 months occurred in HCC patients. The acquisition of resistance to sorafenib is complex, and the contributing mechanism is still unknown [
46]. The limited therapeutic impact of sorafenib and the complex molecular pathophysiology of HCC have made it necessary to conduct novel research projects on sorafenib combinations with other molecular targeting drugs. Sorafenib has been coupled with antiangiogenic drugs, MEK/ERK pathway inhibitors, EGF/EGFR pathway inhibitors, and inhibitors of the HGF/c-Met pathway [
47]. Overall, sorafenib prolongs overall survival by approximately 3 months in patients with aHCC and remains one of the best first-line treatment options with an acceptable tolerability and safety profile.
2.2. Sunitinib
Sunitinib is an orally administered multitargeted tyrosine kinase inhibitor with anticancer and antiangiogenic activity toward VEGFRs, PDGFRs, and several other related tyrosine kinases [
48]. Only one phase III trial (SUN1170) evaluated the drug’s efficacy as a first-line medication for HCC; however, it was terminated because of side effects. In any case, sunitinib appears to be less effective than sorafenib in terms of overall survival (7.9 vs. 10.2 months,
p = 0.0014). Based on existing evidence, sunitinib is not a feasible treatment option as a replacement for sorafenib [
49]. Comparisons between sunitinib and sorafenib demonstrated that sunitinib had a significantly lower overall survival rate, despite no significant difference in progression-free survival.
2.3. Brivanib
Brivanib is a synthetic drug that inhibits both VEGFR and FGFR tyrosine kinase activity [
50]. In the BRISK-FL study, the drug was compared to sorafenib as a first-line treatment, and subsequently to a placebo as a second-line treatment for patients unable to tolerate or respond to sorafenib in the BRISK-PS trial. However, despite similar outcomes in terms of overall survival (time to progression), objective response rate (ORR), and disease control (DC), the intended non-inferiority criterion for overall survival was not met [
51].
Overall survival did not differ significantly between brivanib and sorafenib (9.5 vs. 9.9 months) in the BRISK-FL study and with placebo (9.4 vs. 8.2 months) in the BRISK-PS study. Although currently we cannot consider brivanib as a deserving alternative to sorafenib, better tumor progression times resulting from the BRISK-PS trial and the absence of cross-tolerance with sorafenib have made brivanib an appropriate option for further trials [
52].
2.4. Linifanib
Linifanib is a newly developed ATP-competitive suppressor of all VEGF and PDGF RTKs with no effect on cytosolic TKs or serine/threonine kinases. Due to the high rates of vascularization caused by VEGF overexpression, angiogenic TKIs are thought to be a deserving option in aHCC therapy [
53]. Prior preclinical studies showed some anti-proliferative and pro-apoptotic effects of linifanib against tumor cells by suppressing TKRs such as FLT-3. In an open-label, phase II trial, linifanib monotherapy in patients with advanced HCC resulted in a median TTP of 5.4 months and a median OS of 9.7 months among the trial participants, with 89% of Asian ethnicity, which compared favorably with the corresponding results for patients in the phase III sorafenib study of the Asia-Pacific region [
37,
54]. In a large open-label, randomized Phase III trial (LiGHT) among 1035 patients with advanced or metastatic HCC, results indicated that linifanib was associated with longer TTP (5.4 vs. 4.0 months), higher ORR (13.0% vs. 6.9%), and more frequent adverse events (54% vs. 38%), including grades 3 and 4 hypertension, encephalopathy, ascites, and hyperbilirubinemia. The median OS was 9.1 months for linifanib (95% CI, 8.1 to 10.2) and 9.8 months for sorafenib (95% CI, 8.3 to 11.0), with no significant difference [
55]. Despite similar overall survival and a statistically significant increase in time to progression in the linifanib arm compared to the sorafenib arm, the pre-defined non-inferiority margin for overall survival was not exceeded. Overall, linifanib cannot be considered the best treatment option due to the issues it faces in terms of efficacy and toxicity.
2.5. Lenvatinib
One of the most frequently prescribed drugs in the world, lenvatinib is an orally active inhibitor of multiple receptors of the tyrosine kinase, including VEGFR 1–3, FGFR 1,2–4, PDGFRα [
56], and KIT. Lenvatinib was recently approved as a first-choice therapy for non-excisable HCC (August 2018) [
57].
For more than a decade, sorafenib was the only effective first-line therapy available until Lenvatinib was recently demonstrated to be comparable to sorafenib in terms of overall survival [
28]. For advanced HCC patients, the drug demonstrated promising anticancer activity, with a response rate of 23.9%, a median progression time of 9.4 months, and a survival time of 18.3 months in Phase II studies [
58]. A worldwide Phase III trial (REFLECT) comparing lenvatinib with sorafenib as a first-line treatment was performed as a non-inferiority study based on the outcomes of this Phase II trial [
28]. Among 954 participants, lenvatinib users lived an average of 13.6 months longer, in comparison with 12.3 months for sorafenib users. Overall response rate and progression-free survival were markedly better in the lenvatinib group.
A higher incidence of hypertension, proteinuria, and hepatic encephalopathy was reported in the lenvatinib group. According to the trial’s results, sorafenib was shown to be non-inferior but not greater than lenvatinib when it came to overall survival. Lenvatinib cannot yet completely replace sorafenib as a standard of therapy, despite both medications being deemed standard of care [
59]. Some oncologists may choose sorafenib due to the lesser side effects, while others may pick lenvatinib owing to the antitumor activity or cost-effectiveness. Lenvatinib 12 mg/day costs less than half as much as sorafenib 800 mg/day based on the results of a trial conducted in Japan, since the suggested dosage of lenvatinib (12 mg/day) for patients with advanced HCC is less than half that indicated for cases having other sorts of malignancy, such as thyroid cancer (24 mg/day) [
4]. Administering the most often employed drug in first-line HCC therapy will depend on market conditions following lenvatinib authorization. Lenvatinib was not tested in patients with significant portal vein thrombosis, bile duct invasion, or liver involvement of more than 50% [
60]. Further research on lenvatinib’s effectiveness in combination with other classes of drugs is required. Lenvatinib, an FDA-approved therapy option for HCC, has similar adverse effects as sorafenib, and thus the choice of drug may be based on cost effectiveness for the patient.
2.6. Donafenib
Donafenib, a derivative of sorafenib, is a novel multikinase inhibitor of multiple receptor kinases, including VEGFR, PDGFR, and Raf kinases, leading to suppressed tumoral growth and angiogenesis. Donafenib has been demonstrated to be efficient and safe in some preclinical phase Ia and Ib trials [
61,
62]. In an open-label, randomized phase II/III trial [
63] among the donafenib and sorafenib groups, results showed a significant increase in overall survival (12.1 and 10.3 months, respectively). Although the median time to progression (3.7 months vs. 3.6 months), the objective response rate (4.6% vs. 2.7%), and the disease control rate (30.8% vs. 28.7%) were not significantly different. Donafenib demonstrated better safety and tolerability compared to sorafenib; drug-related AEs of grade ≥ 3 were experienced by 37.5% of patients in the donafenib group, versus 49.7% in the sorafenib group, and the incidence of treatment interruptions caused by drug-related AEs was 30.3% in the donafenib group, compared to 42.5% in the sorafenib group. Overall, donafenib improves OS significantly compared to sorafenib with a better safety and tolerability profile. Therefore, donafenib has the potential to be an effective first-line treatment for advanced HCC; however, further research through larger scale trials is necessary to accurately assess its efficacy and safety.
2.7. Atezolizumab + Bevacizumab
An immune checkpoint inhibitor, atezolizumab, aims for programmed cell death ligand 1 (PD-L1), while bevacizumab is a vascular endothelial growth factor (VEGF) monoclonal antibody. There is emerging evidence to support the application of these two drugs together for the treatment of metastatic HCC [
64].
For healthy individuals with no worse than Child–Turcotte–Pugh class A cirrhosis (
Table 1), an excellent performance status, no contraindications to bevacizumab, and no post-liver transplantation relapse, atezolizumab plus bevacizumab combination therapy is suggested rather than sorafenib monotherapy. This recommendation is consistent with the 2020 guideline from ASCO [
65], the 2021 guidelines from the Society for Immunotherapy of Cancer [
66], and a position paper from the European Association for the Study of the Liver [
22].
Combination therapy was directly compared to sorafenib monotherapy in an open-label phase III trial called IMBrave150 among 501 previously untreated patients with advanced, unresectable HCC who did not have worse than Child–Turcotte–Pugh class A cirrhosis. Atezolizumab (1200 mg intravenous (IV) every three weeks) plus bevacizumab (15 mg/kg IV every three weeks after Atezolizumab) were administered to 336 patients, and sorafenib (400 mg orally twice daily) to 165 patients [
29]. IMbrave150 research indicated that atezolizumab plus bevacizumab outperformed sorafenib with a median OS of 19.2 vs. 13.4 months and an ORR of 29.8% vs. 11.3%, respectively, with a higher rate of complete responses in the combination therapy group (CR = 7.7%). This combination regimen has demonstrated the longest overall survival rate ever seen in first-line phase III studies, further affirming its potential to become the standard of care for patients with advanced HCC who have not received prior systemic therapy. In the case of adverse effects, a similar number of patients in both groups (57 vs. 55 percent) experienced grade 3 or 4 side effects, showing a tolerable safety profile. Although combination therapy was associated with higher rates of hypertension, pyrexia, alanine transaminase elevation, and proteinuria. The FDA has authorized atezolizumab plus bevacizumab for unresectable or advanced metastatic HCC in patients who have not previously completed systemic therapy in light of these findings [
67].
2.8. Sintilimab + Bevacizumab
Sintilimab is an IgG4 monoclonal antibody that boosts T-cell anticancer activity by binding to programmed cell death receptor-1 (PD-1) and suppressing the interaction of PD-1 with its ligands (PD-L1 and PL-L2). Sintilimab was found to outperform sorafenib in the Chinese ORIENT-32 study in combination with IBI305 (bevacizumab biosimilar) [
68]. ORIENT-32 was a randomized, open-label, phase II–III trial conducted at 50 Chinese clinical locations among 595 patients with unresectable HCC. Combination therapy showed a significantly longer overall survival than did sorafenib (median not reached) vs. 10·4 months. In phase II of the study 7 (29%) of the patients experienced grade 3 or worse adverse events during treatment. In phase III, the sintilimab–bevacizumab biosimilar group showed a significantly longer median PFS (4·6 months) than the sorafenib group (2·8 months). The most common grade 3–4 treatment-emergent adverse events in phase III were hypertension and hand-foot syndrome [
69]. Overall, sintilimab plus IBI305 significantly improved overall survival and PFS versus sorafenib in patients with unresectable, HBV-associated HCC with a tolerable safety level. This combination therapy regimen could be an effective option for such patients [
68].
2.9. Cediranib
Cediranib is an orally administered suppressor of RTKs that specially affects vascular endothelial growth factor-A (VEGF-A or VEGF), and it was developed for the purpose of suppressing tumor growth, neovascularization, and metastasis [
70]. A phase II trial was conducted in 2006 among 28 patients with unresectable or metastatic HCC. Patients received 45 mg of cediranib orally, once daily, for 28-day cycles. All 28 patients were evaluable for efficacy outcomes. Twelve patients (42.9%) survived 6 months, 15 (53.6%) died within 6 months, and one (3.6%) was lost to follow-up before 6 months. The median OS was 5.8 months (95% CI: 3.4–7.3 months). No partial or complete response was observed. The median TTP was 2.8 months (95% CI: 2.3–4.4 months). Twenty-six patients (93%) experienced a grade 3+ adverse event (AE), with the most common AEs being fatigue (46%), anorexia (25%), hypertension (21%), and elevated alanine aminotransferase (ALT) (18%). This phase II trial showed stable disease in 25% of the patients treated. However, with respect to the lower tolerance and rate of response compared to sorafenib, further development of cediranib with the dose and schedule used in this trial was not justifiable [
71]. In the SHARP trial, the use of sorafenib resulted in a 71% rate of stable disease [
37]. Another phase II trial was conducted in 2009 among 17 patients with advanced HCC, and sufficient hematologic, hepatic, and renal functions were observed in those who received cediranib 30 mg/d (4 weeks/cycle). With a median follow-up time of 17 months, the median PFS of this cohort was 5.3 months [95% CI: 3.5–9.7 months], and the median OS was 11.7 months [95% CI: 7.5–13.6 months]. The estimated three-month PFS rate was 77% [95% CI: 60–99%] [
71]. However, in the previous phase II study in advanced HCC, the use of cediranib at 45 mg daily led to toxicity in 93% of the patients, including grade 3 or above adverse events, including fatigue (46%), anorexia (25%), and hypertension (21%) [
70]. In this study, a different tolerability profile was observed. Grade 3 events of fatigue and anorexia were low (5% and 0%, respectively). A high incidence of grade 3 toxicities, including hypertension (29%), hyponatremia (29%), and hyperbilirubinemia (18%), was observed. Only modest evidence of antitumor activity (disease stabilization) was found in this small cohort of 17 HCC patients treated with cediranib [
71]. The median PFS (5.3 months) and OS (11.7 months) in this group of patients compared favorably to data reported with 45 mg/d dosing of cediranib in advanced HCC (TTP of 2.8 months and OS of 5.8 months). Overall, cediranib at either a 30 mg or 45 mg daily dosage showed a high incidence of toxicity and preliminary evidence of antitumor activity in advanced HCC and cannot be considered an appropriate option for HCC therapy.
2.10. Nintedanib
Nintedanib is an orally administered triple angiokinase inhibitor of VEGFR1-3, PDGFRα and β, FGFR1-3, Flt-3, Lck, Lyn, and Src, with anti-tumor and anti-angiogenic activity in preclinical models of HCC [
56]. In a phase-II trial, 93 patients were randomized in a 2:1 ratio to receive nintedanib 200 mg bid (n = 62) or sorafenib 400 mg bid (n = 31) continuously in 28-day cycles, until intolerable AEs or disease progression (PD) [
72]. Median TTP was 5.5 vs. 4.6 months, median OS was 11.9 vs. 11.4 months, and median PFS was 5.3 vs. 3.9 months, respectively. Dose intensity and tolerability favored nintedanib. Fewer patients on nintedanib (87.1%) vs. sorafenib (96.8%) had drug-related adverse events (AEs) or grade ≥ 3 AEs (67.7% vs. 90.3%), but more patients on nintedanib (28 (45.2%)) had AEs leading to drug discontinuation compared to those on sorafenib (7 (22.6%)). Approximately a quarter of patients on nintedanib experienced GI AEs of grade 3 or higher. Nausea (48.4% vs. 29.0%), vomiting (38.7% vs. 29.0%), and upper abdominal pain (25.8% vs. 12.9%) occurred >10% more frequently with nintedanib compared with sorafenib. Hand-foot syndrome (35.5% vs. 1.6%), alopecia (35.5% vs. 4.8%) and rash (22.6% vs. 9.7%) were more frequent with sorafenib compared with nintedanib. Overall, nintedanib may have similar efficacy to sorafenib in an HCC with a tolerable and different safety profile but with higher VEGF-related toxicity. The results suggest that nintedanib could be a suitable option for combination studies in HCC.
2.11. Dovitinib
Dovitinib, a strong inhibitor of FGFRs, VEGFRs, and PDGFR, exhibits anticancer efficacy through antiproliferative and antiangiogenic mechanisms [
73]. A phase II randomized trial of Asian-Pacific patients with advanced HCC comparing the clinical activity of dovitinib (500 mg/day, 5 days on, 2 days off; n = 82) vs. sorafenib (400 mg twice daily; n = 83) showed comparable OS and median time to progression. The median overall survival (mOS) was 8.0 months for dovitinib vs. 8.4 months for sorafenib, and the median time to advance on investigator assessment was 4.1 months for dovitinib vs. 4.1 months for sorafenib. Common adverse events included diarrhea (62%), decreased appetite (43%), nausea (41%), vomiting (41%), fatigue (35%), rash (34%), and pyrexia (30%) for dovitinib; hand-foot syndrome (66%) and decreased appetite (31%) for sorafenib [
74,
75]. Another phase II trial among 24 patients with early and intermediate-stage HCC who received neoadjuvant oral dovitinib 500 mg daily (5 days on/2 days off) for 4 weeks after locoregional therapy showed a decrease in intratumoral blood flow and a mild anticancer response. The most frequent grade 3–4 adverse events that emerged in 88% of patients were hypertension (54%), fatigue (25%), and thrombocytopenia (21%) [
76]. Overall, dovitinib may not be a vigorous rival for sorafenib as the frontline systemic drug used for HCC treatment, but it might be used as a systemic neoadjuvant therapy after dose adjustments due to poor tolerability.
2.12. Everolimus
Everolimus is an orally administered mTOR inhibitor [
77]. The mammalian target of rapamycin (mTOR), which is located in the downstream of the PI3K AKT pathway, is important in angiogenesis, cell cycle progression, and proliferation of hepatic tumor cells. Activation of the mTOR pathway is observed in various solid cancers, including 30–40% of HCC [
78]. EVOLVE-1 was a randomized, double-blind, phase III study conducted among 546 adults from 17 countries with BCLC stage B or C HCC whose disease progressed during or after sorafenib or who were intolerant of sorafenib. A total of 362 patients were randomized to the 7.5 mg/d everolimus group and 184 patients to the placebo group. No significant difference in OS was seen between treatment groups, with 303 deaths (83.7%) in the everolimus group and 151 deaths (82.1%) in the placebo group (HR = 1.05; 95% CI, 0.86–1.27;
p = 0.68; median OS, 7.6 months with everolimus, 7.3 months with placebo). The median TTP with everolimus and placebo was 3.0 months and 2.6 months, respectively (HR, 0.93; 95% CI, 0.75–1.15), and the disease control rate was 56.1% and 45.1%, respectively (
p = 0.01). The most common grade 3/4 adverse events for everolimus vs. placebo were anemia (7.8% vs. 3.3%, respectively), asthenia (7.8% vs. 5.5%, respectively), and decreased appetite (6.1% vs. 0.5%, respectively) [
79]. In the SAKK77/08 and SASL29 trials among 106 patients with unresectable or metastatic HCC, comparing the efficacy of (800 mg/d sorafenib) monotherapy against (800 mg/d sorafenib + 5 mg/d everolimus) combination therapy, no evidence was found that combination therapy improves efficacy compared to monotherapy. Even so, it can be more toxic [
80]. In another single-arm phase I/II study among 25 patients with advanced HCC, 10 mg/day of everolimus was administered, and some preliminary antitumor activity was observed [
81]. Grade 3–4 adverse events included lymphopenia (n = 3), aspartate transaminase (n = 3), hyponatremia (n = 2), and 1 patient each with anemia, alanine transaminase, hyperglycemia, proteinuria, rash, and hypoxia. One patient (4%) had a partial response (95% confidence interval [CI], 0.9–19.6%). The median PFS and overall survival were 3.8 months (95% CI, 2.1–4.6) and 8.4 months (95% CI, 3.9–21.1), respectively. The estimated PFS rate at 24 weeks was 28.6% (95% CI, 7.9–49.3%). Overall, everolimus is not able to improve OS in patients with advanced HCC, and it cannot be considered a first-line treatment for such patients, but it can delay tumor progression and can be expected as a second-line therapy in HCC resistant to sorafenib.
2.13. Tislelizumab
The programmed cell death-1/programmed cell death ligand-1 (PD-1/PD-L1) axis plays a central role in suppressing antitumor immunity, and axis dysregulation can be used by cancer cells to evade the immune system [
82]. Tislelizumab, an investigational humanized IgG4 monoclonal antibody with high affinity and binding specificity for PD-1, was engineered to minimize binding to FcγR on macrophages to limit antibody-dependent phagocytosis, a potential mechanism of resistance to anti-PD-1 therapies [
83]. A preliminary report of the HCC cohort consisted of 50 previously treated HCC patients that were treated with tislelizumab 5 mg/kg every 3 weeks, and the most common treatment-emergent AEs were decreased appetite, rash, decreased weight, and cough. Overall, the safety profile of tislelizumab included mostly mild-to-moderate AEs. This preliminary safety profile and antitumor activity support the continued development of tislelizumab as a single agent or in combination with other agents in patients with unresectable HCC [
84]. RATIONALE 301 was a worldwide Phase III open-label, randomized, multicenter trial comparing the effectiveness and safety of tislelizumab as first-line treatment compared to sorafenib in 674 patients with unresectable HCC who had no history of prior systemic therapy. Tislelizumab monotherapy demonstrated clinically meaningful and noninferior overall survival (mOS: 15.9 vs. 14.1 months, respectively), a higher ORR (14.3% vs. 5.4%), more durable responses (mDoR: 36.1 vs. 11.0 months), and fewer grade ≥ 3 adverse effects compared to sorafenib in patients with unresectable HCC. Overall, tislelizumab showed a noteworthy therapeutic advantage over sorafenib with a tolerable and manageable safety profile as a first-line treatment for patients with unresectable HCC [
85].
2.14. Regorafenib
Regorafenib is a multitargeted TKI derived from sorafenib, and its molecular structure is modified by an added fluorine atom in the central phenyl ring of sorafenib. This great change makes Regorafenib stronger in terms of its inhibitory profile and pharmacological activity. Regorafenib influences a variety of tyrosine kinases involved in angiogenesis and tumor growth [
86,
87,
88]. As a second choice of medication following progression despite using sorafenib, regorafenib can be used instead of nivolumab/ipilimumab or pembrolizumab monotherapy. Among 36 cases having advanced hepatocellular carcinoma (HCC), sorafenib was shown to be safe and effective (72.2 percent disease control rate, median time to progression, and median survival of 13.8 months) [
89]. Because of this, a global Phase III trial (RESORCE) comparing regorafenib to placebo was conducted in patients with advanced HCC who developed illness while receiving sorafenib treatment or shortly afterward [
32]. Sorafenib tolerance was defined as using 400 mg/day or more of sorafenib for at least 20 of the previous 28 days of therapy and being in Child-Pugh class A. A total of 573 individuals were randomized and assigned to receive regorafenib or a placebo in a 2:1 ratio. Once a day, one 160 mg dose was given for four weeks, with a one-week break in between the third and fourth weeks. The most critical metric was the capacity to survive. Those who received regorafenib had a median survival time of 10.6 months, while those who received a placebo had a median survival time of 7.8 months, a statistically significant difference (hazard ratio, 0.62;
p < 0.001). Patients in the regorafenib category had a remarkable median survival duration of 26 months after commencing sorafenib [
90]. Overall response rate, duration of progression, and progression-free survival were all significantly different between the two therapy groups. Adverse effects were all similar to the toxicity record of regorafenib, including hand–foot syndrome, weariness, high blood pressure, liver dysfunction, and hypophosphatemia [
91]. As a result of these findings, in April 2017, the FDA extended the eligibility for regorafenib to cover patients previously treated with sorafenib who have been diagnosed with HCC. Regorafenib trials are reasonable for patients who have progressed after receiving first-line sorafenib, who have good performance status and adequate liver function, and who are willing to accept treatment-related morbidity in exchange for the possibility of a small increase in median overall survival, despite the fact that the proper patient classification for second-line regorafenib has not yet been established [
32]. Overall, regorafenib is the only systemic treatment shown to provide a survival benefit in HCC patients progressing on sorafenib treatment.
This entry is adapted from the peer-reviewed paper 10.3390/livers3010011