Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Organic
The synthetic strategies of oxime derivatives participating in radical-type reactions have been rapidly developed in the last few decades. Among them, the N–O bond cleavage of oxime esters leading to formation of nitrogen-centered radicals triggers adjacent C–C bond cleavage to produce carbon-centered free radicals, which has been virtually used in organic synthesis in recent years.
- radical reactions
- oxime ester
- C-C bond cleavage
1. Radical Addition
1.1. Acyl Addition to Alkenes
In 2019, the Wu and Tung group demonstrated a groundbreaking example of acylation/cyclization of Michael acceptors with acyl radicals derived from acyl oxime esters 1 with C–C bond cleavage. Hence, cyclobutyl ketone derivatives 4 and acylated oxindoles 6 were obtained using fac-Ir(ppy)3 as a photosensitizer at room temperature (Scheme 2) [40]. The mechanism studies show that the acyl oxime esters 1 are first reduced by a IrIII catalyst under 3 W blue-light conditions, and then the acyl radicals 7 are produced by C–C bond cleavage. On the one hand, the acyl radical 7 is captured by styrene 3 and oxidized by IrIV to remove a proton to produce enone 8, which is sensitized by IrIII* to further react with another styrene 3 to lead to cross [2 + 2] cyclization and obtain acylated cyclobutanones derivatives 4. Alternatively, acyl radicals 7 could also add to the C–C double bond of the acrylamides 5 to afford structurally useful acylation oxindole skeletons 6.
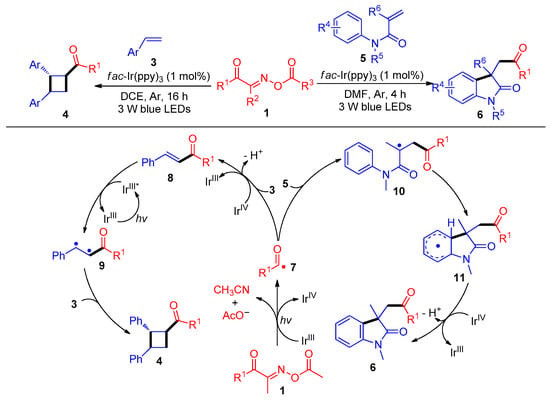
Scheme 2. Visible-light-induced radical acylation/cyclization of acyl oxime esters with olefins.
In the same year, this group reported a novel three-component difunctionalization reaction of acyl oxime esters, olefins and alkane nitriles, which generated a series of β-carbonyl imides 12 with excellent yields (Scheme 3) [41]. The isotope tracking experiment and control experiments revealed the efficient intermolecular reorganization of oxime esters into styrene with the aid of solvent exchange. A possible reaction mechanism is shown in Scheme 3. First, the acyloxime esters 1 are cleaved to nitrile, carboxylate anions 13 and acyl radicals 7 by a single electron transfer (SET) process with the excited state IrIII*, which is generated from IrIII under blue-light irradiation. Then, a SET occurs between intermediate 14, which is produced by the addition of an acyl radical 7 to the olefin 3 and IrIV to complete the cycle of catalyst Ir and produce the carbocation 15. The intermediate 16, produced by the nucleophilic attack of nitrile to cation 15, undergoes a Mumm rearrangement with carboxylate anions 13 to obtain the amide products 12.
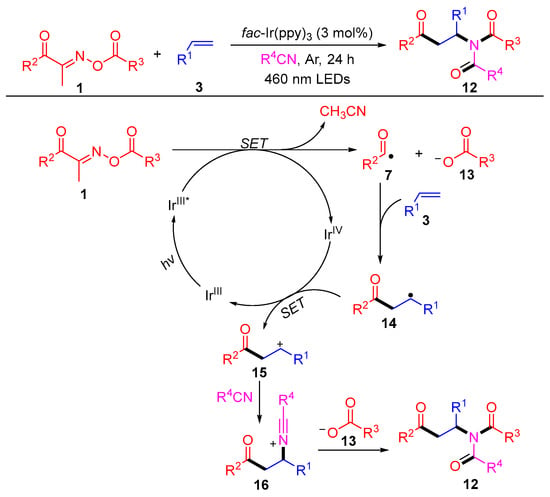
Scheme 3. Visible-light-induced radical difunctionalization of acyl oxime esters with styrenes and nitriles.
A photocatalytic hydroacylation of alkenes with acyl oximes for the synthesis of valuable ketones was developed by Yang et al. (Scheme 4) [42]. When CF3-attached styrenes were used, a E1cB-type fluoride elimination pathway was proposed for the obtainment of the 1,1-difluoroolefin products 19. In this difunctionalization reaction, the triphenylphosphine radical cation generated by the reaction of the excited catalyst IrIII* with triphenyl phosphine combines with the nucleophile acyl oximes 1 to obtain the phosphorus radical intermediate 20. The N–O bond cleavage of the intermediate 20 gives the iminyl radical 21 via β-scission. The imino radical 21 undergoes C–C bond scission to give acyl radical 7 and a molecule of acetonitrile. The acyl radical is added to alkene 17 and reduced by IrII to get the carbanion intermediate 23, with the following protonation to generate the desired product 18.
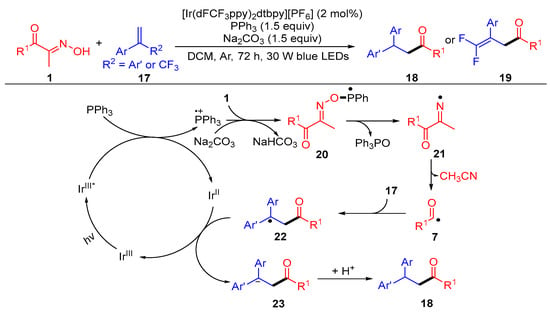
Scheme 4. Visible-light-induced hydroacylation of acyl oximes with alkenes.
In 2021, Sun and co-workers developed a distinctive carbonylation approach using Ir and DIPEA to access γ-keto acids 24 and cyanocarboxylic acids 25 (Scheme 5) [43]. Mechanistically, this reaction involves the photocatalytic radical addition of acyl radicals and cyanoalkyl radicals to aromatic olefins and then carbon dioxide capture. The valuable dicarbofunctionalization tolerates a wide range of cyclic ketoxime substrates; however, only aliphatic acyl oxime esters proceed smoothly to afford corresponding γ-keto ester products.
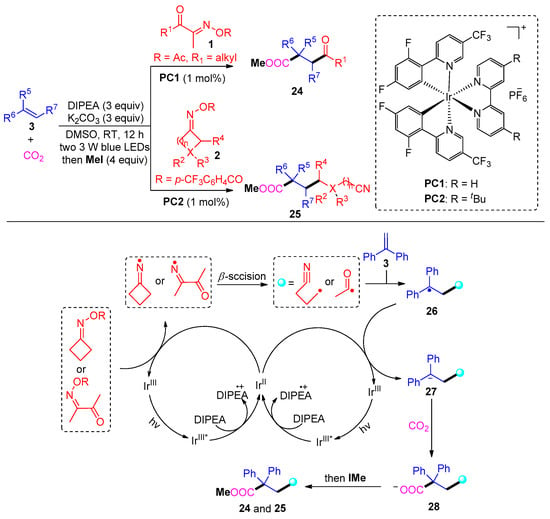
Scheme 5. Visible-light photoredox-catalyzed dicarbofunctionalization of styrenes with oxime esters and CO2.
Larionov’s group described a N-heterocyclic, carbene-photocatalyzed, three-component regioselective 1,2-diacylation of alkenes, using acyl oxime esters and aldehydes as two different acylating agents (Scheme 6) [44]. In particular, the authors demonstrated the mechanism by density functional theory (DFT) and time-dependent-DFT (TD-DFT), where an EDA intermediate is probably formed in the diacylation, which initiates photoexcitation to mediate charge transfer. The EDA intermediate 33, formed by the complexation of the Breslow intermediate 32 with the acyl oxime ester 1, is excited under light conditions and subsequently cleaved to remove BzO- and acetonitrile to give the intermediate 34 and acyl radical 7. The intermediate 14 produced by the addition of the acyl radical 7 to the olefin 3 couples with the intermediate 34 and then decomposes to a diketone product 30 and carbene 31.
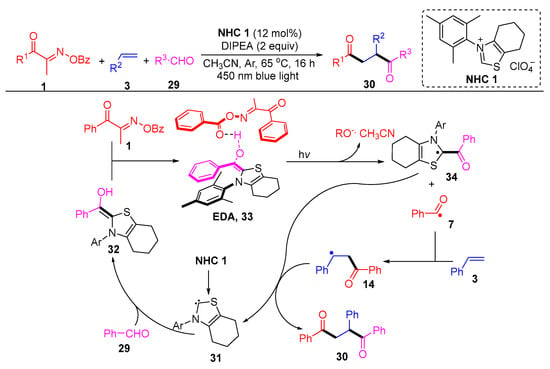
Scheme 6. N-heterocyclic carbene-photocatalyzed 1,2-diacylation of alkenes with acyl oxime esters and aldehydes.
Liu and Huang’s group reported an intriguing route to 3-acyl-substituted chroman-4-one derivatives 36, which involves SET reduction of acyl oxime esters 1 by fac-Ir(ppy)3 under thermal and light conditions (Scheme 7) [45]. In this functionalization reaction, the acyl radical generated from the acyl oxime ester first adds to the carbon double bond of the 2-allyloxy benzaldehydes 35, followed by intramolecular cyclization and 1,2-HAT (hydrogen atom transfer) to obtain the alcohol radical intermediate 40. Finally, SET oxidation by IrIV and deprotonation occur to obtain the target products 36.
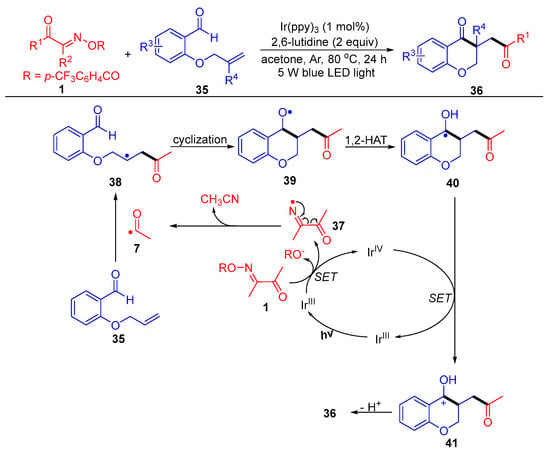
Scheme 7. Visible-light-induced acylation of acyl oxime esters with 2-allyloxy benzaldehydes.
Another acylation/cyclization strategy mediated by a photocatalytic nitrogen-centered radical of acyl oxime esters 1 with activated acrylamides 42 was reported by Liu and Huang’s group (Scheme 8) [46]. The authors screened a variety of acyl oximes with different leaving groups, such as 4-CF3C6H4CO, 4-NO2C6H4CO, C6F5CO, CF3CO, etc., and achieved up to 95% yields with 58 synthetic examples of acylated transformations. The fragmentation of one C–C bond and one N–O bond and the formulation of two new C–C bonds were carried out in one-pot conditions with a low catalyst loading (1 mol%). This protocol represents a simple and green road for the synthesis of acylated indolo/benzimidazo-[2,1,a]isoquinolin-6(5H) ones.
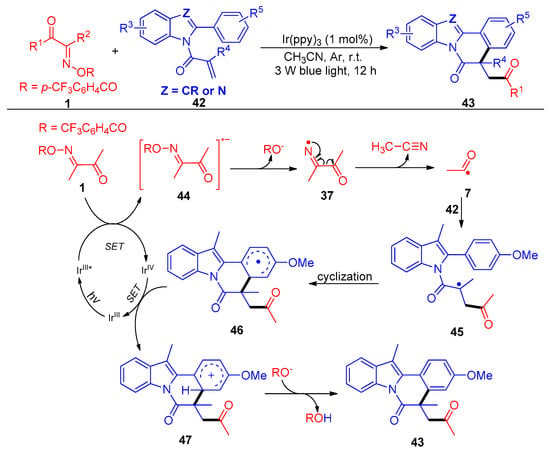
Scheme 8. Visible-light-induced acylation of acyl oxime esters with reactive olefins.
Most recently, the same group uncovered a new acylation reaction of acyl oxime esters with activated N-sulfonyl acrylamides 48, with a broad substrate scope and good functional group compatibility (Scheme 9) [47]. This method provides an effective nitrogen center-mediated approach for the generation of acyl radical intermediates to access acylated oxindole 6. This acylation transformation proceeds through a normal Smiles rearrangement process that involves cascade intramolecular ispo-cyclization (50), de-SO2 (51) and re-cyclization (52).
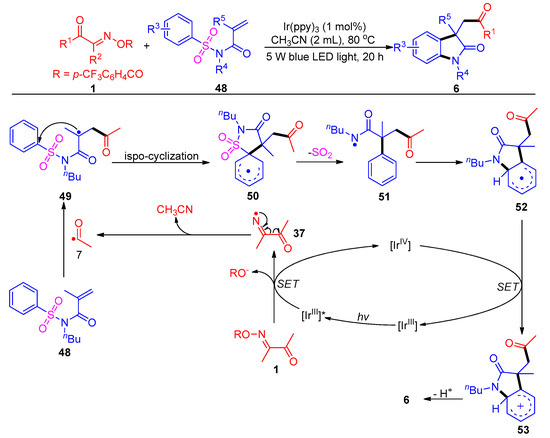
Scheme 9. Visible-light-induced acylation of acyl oxime esters with N-(arylsulfonyl)acrylamides.
1.2. Acyl Addition to Alkynes
The heterocyclic skeleton presents in commercial drugs and many naturally occurring compounds; thus, the development of convenient and environmentally friendly methods for the construction of this structural motif is an important and promising research field [48,49,50]. Alkynes with specific substituents, such as alkyne amides [51,52,53], N-propylindoles [54,55,56], alkyne esters [57,58], alkyne amines [59,60,61], etc., are often used as radical acceptors to construct potential heterocyclic compounds by tandem cyclization. Recently, Liu and Zhou have independently developed a multitude of innovative photocatalytic, nitrogen-centered, radical-mediated acylation strategies of reactive alkynes using acyl oxime esters as acyl sources (Scheme 10) [37,39,62,63].
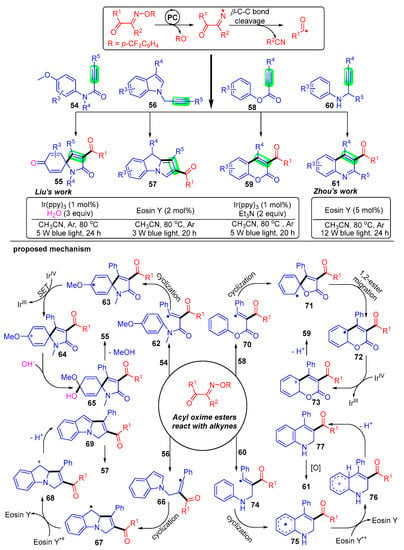
Scheme 10. Visible-light photoredox-catalyzed acylation of alkynes with acyl oxime esters.
Liu’s group pioneered a radical addition strategy of acyl oxime esters 1 with activated alkynes 54 to construct a series of 3-acylated spiro [4,5]trienones derivatives 55 [37]. The acyl radical generated by the C–C bond breakage of acyl oxime esters attacks the carbon–carbon triple bond of propiolamides 54 to give the radical intermediate 62. The intermediate 62 undergoes intramolecular ispo-cyclization and is oxidized to carbocations 64 by IrIV, followed by the combination with hydroxide anion produced in water to obtain the intermediate 65. Finally, a methoxy anion and hydrogen ion are removed to obtain the expected trienone product 55.
Subsequently, Liu’s group developed two pragmatic acylation strategies using N-propynylindoles 56 [63] and alkynoates 58 [62] under photoexcitation conditions. A large quantity of acylated pyrrolo[1,2-a]indole 57 and coumarin 59 derivatives were constructed, and interestingly, both the alkanoyl and aryl groups were well adapted. These studies further developed the application of acyl oxime esters and were of great importance for the development of free radical synthetic chemistry.
Comparably, Zhou’s group has achieved a series of constructions of 3-acyl quinoline skeleton 61 under photocatalytic conditions with N-propargyl aromatic amines 60 as radical acceptors [39]. The mechanism of this acylation reaction states that acyl radicals add to the carbon–carbon triple bond of N-propargyl aromatic amines 60 for intramolecular cyclization and then proceed to dehydroarylation to give the final quinoline product 61.
2. Radical Cross-Couple
Recently, the development of the photoredox/palladium-catalyzed C-H acylation of 2-arylpyridines 78 with acyl oxime esters 1 using fac-Ir(ppy)3 as a photosensitizer was reported by the Chen group (Scheme 11) [64]. In order to thoroughly study the reaction mechanism, the author carried out some control experiments, including the radical clock reaction using benzocycloketoxime ester 80 with ethene-1,1-diyldibenzene under standard conditions and TEMPO as a radical inhibitor to test the activity of the reaction. The generation of the target product 83 and the detection of the free radical capture product 84 by the HRMS indicate that this acylation reaction contains a free radical mechanism.
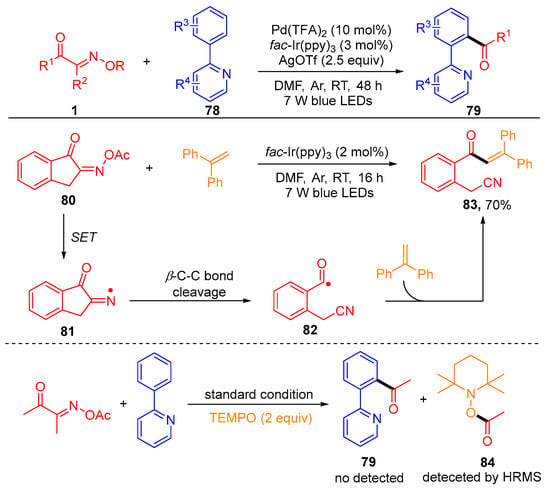
Scheme 11. Photoredox/palladium-catalyzed C-H acylation of 2-arylpyridines with acyl oxime esters.
In addition to the above acyl oxime esters, Wu’s group reported the benzyl oxime esters 85 with the same reaction mode, i.e., a single electron transfer followed by carbon–carbon bond cleavage to produce the corresponding radical (Scheme 12) [38]. The resulting benzyl radicals 91 are further oxidized to benzyl carbocation 92, which are then coupled with O and N-nucleophilic reagents to access the target benzyl ethers 88 and benzylamines 89. This benzylation strategy well tolerates various functional groups under mild conditions, where a wide range of nucleophilic substrates, such as primary and secondary alcohols, amines and even H2O, worked smoothly.
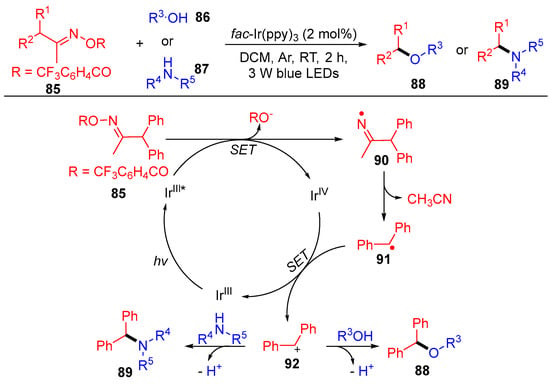
Scheme 12. Visible-light-induced coupling of benzyl oxime esters with O- and N- nucleophiles.
This entry is adapted from the peer-reviewed paper 10.3390/molecules28062667
This entry is offline, you can click here to edit this entry!