Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Mucopolysaccharidosis (MPS) consists of a group of inherited lysosomal storage disorders that are caused by a defect of certain enzymes that participate in the metabolism of glycosaminoglycans (GAGs). The abnormal accumulation of GAGs leads to progressive dysfunctions in various tissues and organs during childhood, contributing to premature death.
- glycosaminoglycans
- inhibition
- mucopolysaccharidosis
1. Introduction
Lysosomal storage diseases (LSDs) are a large group of over seventy metabolic disorders such as Pompe disease, Gaucher disease, Fabry disease, the Niemann–Pick disorders, and mucopolysaccharidosis (MPS) and are caused by inherited gene mutations that alter lysosomal homeostasis [1]. Lysosomal enzymes are affected the most, and deficiency in them results in a progressive accumulation of specific macromolecules inside the endosomal–autophagic–lysosomal system.
MPS is a group of seven inborn genetic disorders and is characterized by an inherent deficiency of lysosomal enzymes that are responsible for the breakdown of specific glycosaminoglycans (GAGs). The abnormal storage process leads to a broad spectrum of adverse health outcomes depending on GAGs levels and location, contributing progressively to morbidity and early mortality.
Over the past two decades, numerous studies have demonstrated a beneficial effect in the existing treatments for LSDs, including MPS [2]. Nevertheless, despite considerable success in reducing morbidity and improving the quality of life of some MPS patients, current therapies are unable to cure all clinical manifestations such as neurological, skeletal, and cardiorespiratory symptoms. Future treatment options such as targeted gene therapy (TGT), anti-inflammatory therapy, and substrate reduction therapy are currently under experimental stages and their outcomes need to be validated in human trials [3].
MPS pathophysiology emanates not only from the direct effects of elevated GAG storage but also is the result of a complex cascade of secondary events in cells with an intricate interplay, contributing to the dysfunction of affected tissues and the complexity of MPS. Thus, exploring new treatments based on the molecular mechanisms and pathological changes underlying MPS is imperative. Lysosomes are acidic subcellular compartments that play a central role in the turnover and recycling of diverse substrates (e.g., endocytosis products, macromolecules from damaged cells organelles). Beside lipases, glycosidases, nucleases, sulfatases or phosphatases, and several families of proteases, including cysteine cathepsins, are found in lysosomes. Cysteine cathepsins were reported to play a key role for several physiological functions. Accordingly, a dysregulation of their expression and/or their proteolytic activity can lead to the development of various human pathologies, including MPS [4][5]. Considering their role in pathological processes, inhibition of specific cathepsins has become an attractive therapeutic strategy. In addition, the activity of cysteine cathepsins can be controlled in several ways, GAGs (depending on their nature and levels) being the foremost one for some cathepsins [6].
2. Mucopolysaccharidosis
2.1. Incidence, Clinical Features, and Diagnosis
MPS represents a group of rare and inherited lysosomal storage diseases that are clinically heterogeneous and characterized by multiorgan involvement due to the accumulation of mucopolysaccharides, aka glycosaminoglycans (GAGs), at the lysosomal level, resulting in reduced life expectancy. The enzymes involved in the degradation of GAG, including heparan sulfate (HS), dermatan sulfate (DS), chondroitin sulfate (CS), keratan sulfate (KS), and/or hyaluronic acid (HA), singly or in combination, are deficient (absence or malfunction) in these pathologies.
MPS are orphan diseases with an incidence estimated to range from 1 per 25,000 and 1 per 100,000 live births, depending on the MPS type [7]. The incidence of MPS types may also be related to continent and ethnic background [8]. The first cases of MPS were described by Charles Hunter in 1917 [9], and two years later, MPS-I cases were reported by Hurler. MPS-IX is the rarest form of mucopolysaccharidosis, with only four patients diagnosed to date [10]. MPS are autosomal recessive genetic diseases, except for MPS type II, which is an X-linked genetic disease [11]. Consequently, lysosomal GAGs accumulate progressively in various tissues, and partially degraded GAGs are excreted in urine. Abnormal GAG storage triggers a cascade of cellular events and progressively prompts organ dysfunction. Clinical features depend on the specific enzyme deficiency and the organs affected by GAGs. While not visible at birth, the first clinical symptoms appear during early childhood. Clinical symptoms are mainly coarse facial features, connective and bone damage, cardiac, respiratory, hearing, and vision disorders, and in most cases, mental retardation [12]. Symptoms may be similar or vary among the different MPS. Clinical examination and several qualitative and quantitative tests (e.g., Elisa, dye-spectrometric, thin layer chromatography, electrophoresis, LC-MS/MS methods) to evaluate GAG levels in urine are the first steps in the diagnosis of a MPS disease [13][14][15][16][17].
2.2. Management
At present, there is no effective curative treatment that can restore mutated genes of patients with MPS. However, depending on the degree of severity and timely diagnosis, different therapeutic options are possible. Current clinical practices such as HSCT and ERT are dedicated to mitigating the progression of MPS and improving the quality of life of patients [18]. Novel experimental therapies for MPS such as gene therapy (GT), anti-inflammatory therapy, substrate reduction therapy (SRT), and pharmacological chaperone therapy have been investigated and may represent promising avenues.
The understanding of mucopolysaccharidosis has been facilitated by the study of animal models, which naturally present the same phenotypes as humans due to mutations in orthologous genes. These animals are often domestic species, especially dogs and cats. Over the past few decades, knockout mouse models with phenotypes similar to the different types of MPS have emerged [19].
2.2.1. Hematopoietic Stem Cell Transplantation
HSCT involves a blood cell transplant of donor cells from three different sources: bone marrow, peripheral blood stem cells, and umbilical cord blood (for review: [20]). The monocyte–macrophage system is the basic mechanism of therapeutic action, as it relies on the ability of circulating monocytes to escape from vessels and migrate inside organs where they transform into macrophages. When macrophages reach the different sites, they secrete the functional enzyme, which is internalized by the surrounding affected cells; the enzyme then reaches the lysosomes and degrades the stored and undigested material. However, as this process is slow and incomplete, success is limited for the treatment of severe neurological diseases. Although very few studies have used this approach, HSCT has been shown to increase life expectancy and improve clinical manifestation in children with attenuated Hurler disease when performed early in life and with preparative conditioning regimens to reduce graft–host disease, infection, and additional complications [21]. For other types of MPS, HSCT has not had the same success as for Hurler’s disease, and other therapeutic approaches have been developed.
2.2.2. Enzyme Replacement Therapy
In 1964, Christian de Duve first suggested that ERT might be a therapeutic option to treat lysosomal storage diseases [22]. ERT has been approved for MPS-I (2003), MPS-II (2006), MPS-IVA (2015), MPS-VI (2005), and MPS-VII (2017). The aim of ERT is to compensate metabolic defects in MPS patients by weekly or fortnightly infusions of recombinant enzymes. Contrary to HSCT, in which the functional enzyme is expressed and circulated indefinitely, enzyme experiences in ERT display a rapid clearance and a half-life, typically under 1 h. The clinical efficacy is highly variable depending on the health status of the patients. Moreover, it is very difficult to target the recombinant enzymes on tissues that are not easily accessible by the systemic circulation, in particular bones, cartilages, or brain (blood–brain barrier), although the search for new and more effective therapeutic strategies is in progress [23][24][25][26]. Alternatively, the use of ERT in combination with HSCT may present significant therapeutic benefits by the possibility to resolve immune response and reduce symptoms and decrease mortality rates [20].
2.2.3. Gene Therapy
The success of this approach has been demonstrated in several MPS animal models [27]. Phase I/II clinical trials are underway for MPS-I, -II, -IIIA, -IIIB, and -VI in several countries (for review: [28]). It involves either in vivo therapy, with the direct injection of therapeutic gene intravenously or locally to target somatic cells through an appropriate viral or non-viral vector, and ex vivo therapy, in which the vector is transfected into somatic cells derived from MPS patient and then re-administrated into the recipient. Transduced cells should be able to continuously secrete supra-physiological enzyme levels in all organs affected by MPS. As the secreted enzyme cannot cross the blood–brain barrier, the benefits of this approach are generally limited to peripheral organs, although recent studies have also shown the efficiency of gene transmission and expression after injection directly into the central nervous system (CNS) (for review: [29]). A phase I/II clinical trial has been launched in patients with type IIIA MPS [30]. The delivery of sufficient enzyme in CNS and bone, the high immunogenic toxicity of both vectors and transgene, and the relatively high cost of this technology remain an unmet challenge for GT [31].
2.2.4. Anti-Inflammatory Drugs, Substrate Reduction, and Pharmacological Chaperone Therapeutic Strategies
In the last decade, new therapeutic options have been investigated for MPS patients and are under clinical trials such as anti-inflammatory drugs, substrate reduction therapy (SRT), and pharmacological chaperone therapy. To suppress metabolic inflammation caused by GAGs accumulation, anti-inflammatory treatments in combination with current MPS treatment could be an alternative to inhibit secreted cytokines using blocking antibodies, impair cell–cell interactions, or suppress specific cell types [32][33][34]. Substrate reduction therapy (SRT) is another therapeutic approach, which consists of directly or indirectly slowing down GAG biosynthesis with the use of small inhibitors to reduce lysosomal storage. Contrary to ERT, these small molecules can cross the blood–brain barrier and have the potential to directly treat CNS symptoms of MPS. Preclinical and clinical trials, however, showed various outcomes [35][36], and SRT has not been approved yet for any MPS. In MPS, deficiencies in enzymes involved in GAG catabolism are due to mutations, which in some cases affect full processing, folding, and lysosomal targeting. Pharmacological chaperone therapy (PCT) aims to use small molecules that specifically bind to the mutated enzyme to enhance its correct folding, stability, and intracellular trafficking [37]. PCTs have the advantage of wide tissue distribution, potential oral distribution, and low immunogenicity.
3. Glycosaminoglycans in MPS
GAGs are a family of highly complex, linear, and heterogeneous polysaccharides that consist of repeating disaccharide units with varying chain length, type of linkage, and extent of sulfation and epimerization. They can be categorized into four main groups: heparin (Hep)/heparan sulfate (HS); chondroitin sulfate (CS)/dermatan sulfate (DS); keratan sulfate (KS); and hyaluronan (HA) [38].
3.1. Structure, Expression, Catabolism, and MPS Disorders
GAGs are negatively charged polysaccharide chains with a molecular weight of approximately 10–100 kDa, except for HA, which exhibits molecular weights in the range of 4–8000 kDa. Among GAGs, two categories: non-sulfated (HA) and sulfated GAGs (CS, DS, KS, Hep, and HS) can be distinguished. The chains of GAG are composed of repeated disaccharide units, including uronic acid and a hexosamine, except for KS where uronic acid is replaced by galactose (Gal). Uronic acid exists in two forms: iduronic acid (IdoA) or glucuronic acid (GlcA). For hexosamine, it can be either N-acetyl glucosamine (GlcNAc) or N-acetyl galactosamine (GalNAc) [39]. The structural diversity of GAGs is enhanced by different degrees of modification of the disaccharide subunits. Indeed, the hydroxyl groups in position C2 of uronic acid and in positions C3, C4, and C6 of hexosamine can be O-sulphated, and glucosamines can be N-acetylated or N-sulfated (or more rarely N-unsubstituted). Subsequently, an octa-saccharide could exhibit over 1,000,000 different sulfation sequences [40].
3.1.1. Heparin/Heparan Sulfate
Heparin typically consists of shorter disaccharide repeating units of β1,4-linked α-L-iduronic and α-D-glucosamine, in which the predominant substitution pattern is 2-O-sulfation of the iduronate residues and N- and 6-O-sulfation of the glucosamine residues [41]. Other substitutions including N-acetylation and 3-O-sulfation may be present in glucosamine. In heparan sulfate (HS), uronic acid is predominantly β-D-glucuronic acid, the C5 epimer of α-L-iduronic. HS is naturally present in all cells and varies in terms of degree of sulfation and chain length depending on the biological origin. HS chains are generally made up of 50 to 250 disaccharide units (20 to 100 kDa). At physiological pH, all carboxylic and sulfate functions are deprotonated, giving GAGs high negative charge densities (heparin has the highest negative charge density of any known mammalian GAGs) [42]. Sulfation of the various hydroxyl groups or the amino group present on the glucosamine compound of HS/Hep drives its ability to interact with various proteins, cytokines, and growth factors [43]. While Hep is largely restricted to mast cells, HS is ubiquitously expressed on cell surfaces and in the extracellular matrix (ECM) and basement membrane (BM) in mammalian tissues. HS/Hep are tethered to proteins through a tetra-saccharide linker, covalently bound to a serine residue to form proteoglycans (PGs) (for review: [44]). Heparan sulfate proteoglycans (HSPGs) are classified into three groups according to their location: (i) transmembrane HSPGs, such as syndecans 1–4 (carrying HS and CS chains) and glypicans 1–6 (HS chains), (ii) pericellular and extracellular HSPGs including agrin (HS chains), perlecan (HS chains) and type XV and XVIII collagens (HS chains), and testicans 1–3 (HS chains), and (iii) the secretory vesicle serglycin (Hep and CS chains). Proteoglycans participate in many biological processes such as cell regulation (growth, proliferation, and migration) [45][46], CNS development and repair [47][48], and cell recognition [49][50]. Hurler syndrome, the severe form of MPS-I, is associated with neurological and/or behavioral abnormalities, as observed in MPS-II, -III, and -VII, where HS is accumulated (Figure 1). Since HS is the primary storage material in these MPS types, HS could be an interesting candidate as a biomarker of brain pathology and neurological manifestations for MPS-I, -II, -III, and -VII, by measuring its levels in urine and blood. Treatment of MPS-II mice with a blood–brain-barrier-penetrable antibody (Pabinafusp Alfa) reduces HS levels in brain and prevents neurodegeneration and neurocognitive dysfunction [51]. The accumulation of HS in MPS-I, -II, -III, and -VII affects lysosomal functions, leading to numerous irreversible alterations within and outside cells (e.g., abnormal composition of membranes, intracellular vesicle trafficking, autophagy, mitochondrial dysfunction, oxidative stress, inflammation) [52][53].
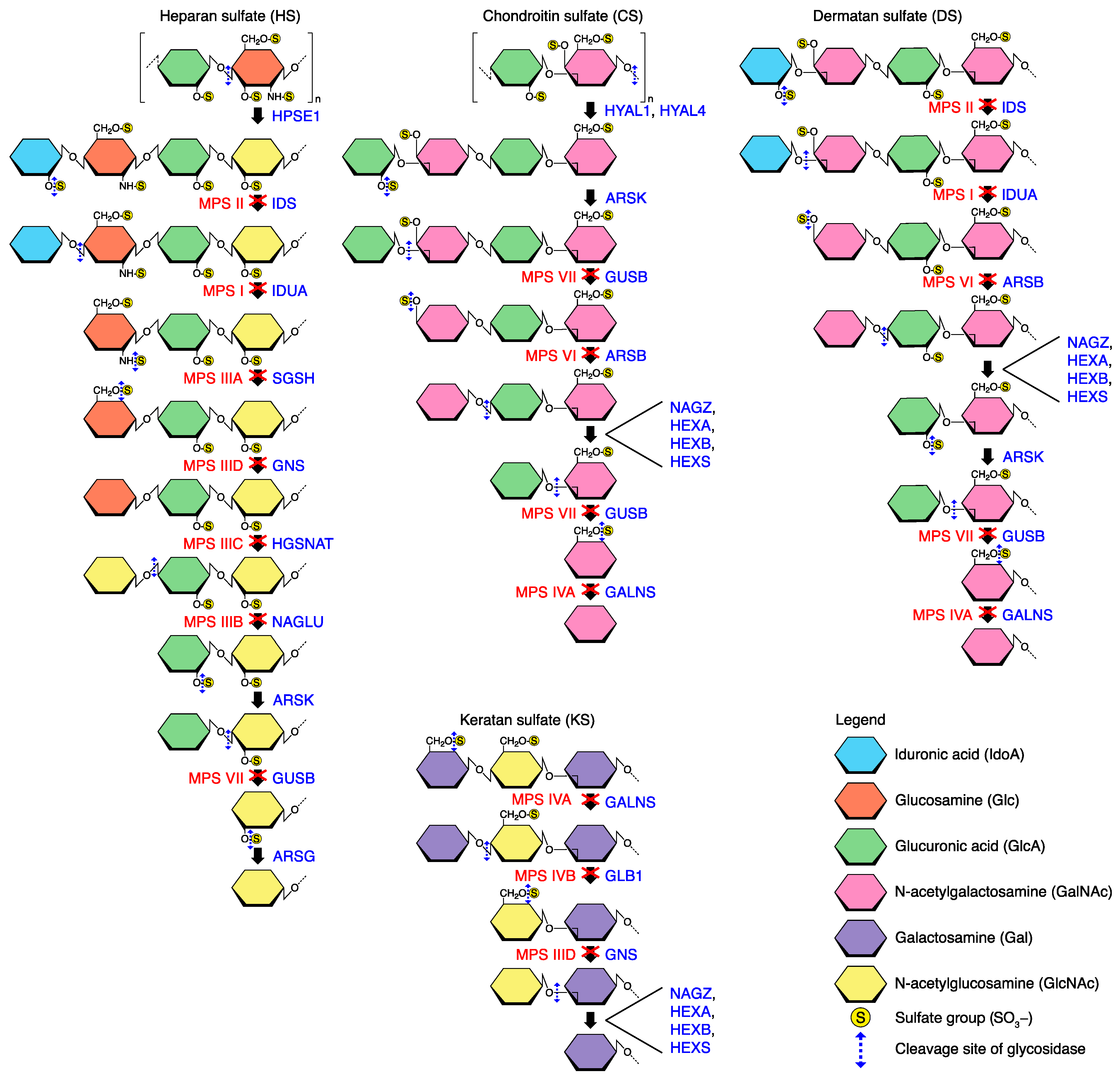
Figure 1. Degradation of the sulfated GAG chains. The deficient enzymes involved in the different MPS subtypes are indicated. HYAL1/4 and HPSE1 are endoglycosydases capable of degrading long chains of CS and HS, respectively, into smaller fragments. Degradation of GAGs consists in repeating steps of desulfation and deglycosylation at the non-reducing end of the chain by sulfatase and exoglycosydase, respectively. Enzymes involved in the catabolism of GAGs and in MPS subtypes associated are indicated. ARSB (arylsulfatase B); ARSG (arylsulfatase G); ARSK (arylsulfatase K); GALNS (galactosamine N-acetyl)-6-sulfatase); GLB1 (β1-galactosidase); GNS (glucosamine (N-acetyl-6-sulfatase)); GUSB (β-glucuronidase); HEXA (hexosaminidase subunit α); HEXB (β-hexosaminidase); HEXS (hexosaminidase S); HGSNAT (heparan-α-glucosaminide N-acetyltransferase); HPSE1 (heparanase); HYAL1/4 (hyaluronidase 1/4); IDS (iduronate 2-sulfatase); IDUA (α-L-iduronidase); NAGLU (N-acetyl-α-glucosaminidase); NAGZ (β-N-acetylglucosaminidase); SGSH (N-sulfoglucosamine sulfohydrolase).
Additionally, accumulation of membrane-bound cell-surface HSPGs may alter growth-factor–receptor interactions and signal transduction [54]. Besides neural dysfunctions, HS can lead to the progressive development of a variety of clinical manifestations, including ear, nose, throat, and respiratory problems, which are often the first emerging symptoms in all MPS types [17][55]. Thickened depositions/secretions in airways and interstitium due to an abnormal accumulation of ECM components can further exacerbate lung obstruction. Due to the unsuitability to monitor respiratory function in young MPS patients, an alternative non-invasive method named global respiratory symptoms severity (GRSS) was developed [56]. GRSS is a score ranging from 0 to 4, which relies on four lung restriction subtypes: (i) ear–nose–throat symptoms (chronic rhinitis or sinusitis, otitis, adeno-tonsillar hypertrophy, hearing loss, macroglossia, stridor), (ii) pulmonary symptoms (dyspnea, wheezing, cough, sputum, asthma, bronchitis, pneumonia), (iii) clinical symptoms of obstructive sleep apnea, and (iv) skeletal abnormalities causing restrictive lung disease (scoliosis, kyphosis, ribcage narrowing, chest wall deformity). Researchers reported that HS levels, which is the most abundant GAG in the lungs, increased in respiratory secretions of MPS-I, -II, and -III young patients compared to non-MPS patients, and correlated positively to the severity of respiratory symptoms (GRSS) that worsen with age [56].
3.1.2. Chondroitin Sulfate/Dermatan Sulfate
Chondroitin sulfate consists of repeating GlcA-GalNAc disaccharide units linked by β1,3 and β1,4, respectively. Sulfate groups can be either present at the C4 (C4-S or CS-A) or C6 (C6-S or CS-C), or both C4 and C6 (C4,6-S or CS-E) hydroxyl groups of GalNAc units. The GlcA unit can also be sulfated at the C2 position, giving rise to CS-D (6-sulfated GalNAc and 2-sulfated GlcA). Although found throughout the body, CS is a major component of ECM (i.e., bone, cartilage, and central nervous system) and is an essential component of PGs (CSPGs) such as aggrecan, versican, and neurocan. The different sulfation patterns confer different roles to CS and allow selective interactions via electrostatic interactions, with positively charged platelet-derived growth factors (PDGFs) fibroblast growth factor (FGF), insulin-like growth factor (IGF), vascular endothelial growth factor (VEGF), and TGF-β, resulting in the stabilization of these growth factors in solution [57]. CS participates in tissue remodeling and homeostasis and exerts anti-inflammatory activity in articular tissues by reducing proinflammatory factors [58]. Overexpression of CS contributes to chronic inflammatory diseases, including skin lupus erythematosus and dermatomyositis or pulmonary fibrotic diseases [59][60]. Dermatan sulfate (DS) is a stereoisomer of CS and formerly named CS-B. DS chains consist of alternating IdoUA-GalNAc units with 50–200 repeats. Sulfation occurs at the C2 and C4 on IdoUA and C6 on GalNAc residues, respectively. The presence of IdoA residue in DS, like in HS and Hep, appears to play a key role in GAG-binding proteins and particularly with chemokines and cytokines, including IL-8, macrophages inflammatory peptides (MIP-1α and β), RANTES (regulated on activation of normal T cell expressed and secreted), and IFN-γ (for review: [61]). DS interacts as well with several other molecules such as growth factors (FGF family), heparin cofactor II, and ECM components. DS is expressed ubiquitously in ECM and is linked to core proteins (DSPGs) such as decorin, biglycan, versican, thrombomodulin, and endocan. DS has a physiological role in anti-coagulation, wound healing, and tissue development but also participates in pathological processes such tumorigenesis and infection [62]. Maroteaux–Lamy syndrome (MPS-VI) is characterized by a deficiency of N-acetylgalactosamine-4-sulfatase that results in the storage of DS and C4-S [63] (Figure 1). Skeleton, bone, and joints are commonly affected. Other progressive somatic deteriorations are reported with age, like in Hurler syndrome, with the exception that the CNS is spared, as HS is not elevated. Features include coarse facies, enlarged tongue, and corneal clouding, among other features. In the severe form of the disease, MPS-VI patients mostly die before the second decade of life due to cardiac and valvular diseases, pulmonary infection, or restrictive lung diseases. Heart disease and airway obstruction are also major causes of early death in MPS-VII patients, following HS, DS, and CS accumulation [64].
3.1.3. Keratan Sulfate
Keratan sulfate is a β-1,4-linked Gal and N-GalNAc, with sulfate residues can be found on the 6-positions of both residues. KS is the only GAG type without acidic residue. KS is found in cornea (the richest source of KS in the human body), tendon, cartilage, bone, and peripheral nervous systems (for review: [65]). Like HS and DS, KS participates in tissue hydration, cellular recognition of protein ligands, and cell motility. There are two forms of KS (KS I and II), depending on the nature of their linkage to protein [66]. KS chains are generally found structurally attached to a protein core forming proteoglycans (KSPGs) including lumican, keratocan, mimecan, osteomodulin, osteoadherin, and fibromodulin. In cornea, the high abundance of KS appears to play a pivotal role in matrix assembly, which is involved in vision acuity [67]. KS, a major component with CS of aggrecan, is also important for maintaining the proper hydration levels in skeletal tissues, conferring resistance to mechanical stress. Other KSPGs (e.g., ABAKAN, claustrin, PG-1000, phosphocan-KS) are present in neural tissues and interact with several nerve regulatory proteins, suggesting the potential role of KS in axonal guidance and neural angiogenic processes [68]. In MPS-IVA/B (Morquio syndrome), deficiency in the galactose 6 sulfate sulfatase (GALNS) and/or β-galactosidase (Figure 1) impairs the further steps of KS catabolism, which results in abnormal KS and C6-S levels in tissues [69][70]. KS concentrations correlated with clinical severity; in particular, KS accumulation in chondrocytes leads to a systemic skeletal dysplasia [71]. Extra-skeletal manifestations include respiratory impairment, sleep apnea, tracheal obstruction/narrowing, hepatomegaly, heart valve disease, hearing loss, corneal clouding, and dental hypoplasia. Although there can be cervical spinal cord compression, abnormal cognitive development is not affected in most MPS-IV cases, contrary to MPS-I, -II, -III, and -VII.
Recently, the six mammalian GAGs (i.e., Hep/HS, CS, DS, KS, and HA) have been reported to bind to more than 800 proteins [72]. While Hep and HS, which are the most extensively studied GAGs, interact with many of the proteins (580), followed by KS and CS with 218 and 72 proteins, respectively, a few ligands bind to HA and DS (43 and 19, respectively). Accumulated GAGs in brain, bone, cartilage, and ECM induces pro-inflammatory factors (e.g., TNF-α, RANTES, IL-1, 2, 5), which leads to the dysregulation of several molecules, including degradative proteases (e.g., MMPs, serine proteases, and cysteine cathepsins), and subsequently to chronic disorders [73]. In addition, GAGs are known to play a key role in the regulation of cysteine cathepsins with diverse effects particularly in in the folding, stability, and activity of proteases. These proteases have received much attention for their diverse roles in physiological and pathological processes, and some of them are very attractive molecular targets for therapeutic interventions (extensively reviewed in [5][74][75][76][77]).
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11030810
References
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal Storage Diseases. Nat. Rev. Dis. Prim. 2018, 4, 27.
- Beck, M. Treatment Strategies for Lysosomal Storage Disorders. Dev. Med. Child Neurol. 2018, 60, 13–18.
- Kobayashi, M.; Ohashi, T.; Kaneshiro, E.; Higuchi, T.; Ida, H. Mutation Spectrum of α-Galactosidase Gene in Japanese Patients with Fabry Disease. J. Hum. Genet. 2019, 64, 695–699.
- De Pasquale, V.; Moles, A.; Pavone, L.M. Cathepsins in the Pathophysiology of Mucopolysaccharidoses: New Perspectives for Therapy. Cells 2020, 9, 979.
- Biasizzo, M.; Javoršek, U.; Vidak, E.; Zarić, M.; Turk, B. Cysteine Cathepsins: A Long and Winding Road towards Clinics. Mol. Aspects Med. 2022, 88, 101150.
- Novinec, M.; Lenarčič, B.; Turk, B. Cysteine Cathepsin Activity Regulation by Glycosaminoglycans. Biomed. Res. Int. 2014, 2014, 309718.
- Çelik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of Mucopolysaccharidoses Update. Diagnostics 2021, 11, 273.
- Zhou, J.; Lin, J.; Leung, W.T.; Wang, L. A Basic Understanding of Mucopolysaccharidosis: Incidence, Clinical Features, Diagnosis, and Management. Intractable Rare Dis. Res. 2020, 9, 1–9.
- Hunter, C. A Rare Disease in Two Brothers. Proc. R. Soc. Med. 1917, 10, 104–116.
- Kiykim, E.; Barut, K.; Cansever, M.S.; Zeybek, C.A.; Zubarioglu, T.; Aydin, A.; Kasapcopur, O. Screening Mucopolysaccharidosis Type IX in Patients with Juvenile Idiopathic Arthritis. JIMD Rep. 2016, 25, 21–24.
- Muenzer, J.; Beck, M.; Giugliani, R.; Suzuki, Y.; Tylki-Szymanska, A.; Valayannopoulos, V.; Vellodi, A.; Wraith, J.E. Idursulfase Treatment of Hunter Syndrome in Children Younger than 6 Years: Results from the Hunter Outcome Survey. Genet. Med. 2011, 13, 102–109.
- Neufeld, E.F.; Muenzer, J. The Mucopolysaccharidoses. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2019.
- Khan, S.A.; Mason, R.W.; Giugliani, R.; Orii, K.; Fukao, T.; Suzuki, Y.; Yamaguchi, S.; Kobayashi, H.; Orii, T.; Tomatsu, S. Glycosaminoglycans Analysis in Blood and Urine of Patients with Mucopolysaccharidosis. Mol. Genet. Metab. 2018, 125, 44–52.
- de Jong, J.G.; Wevers, R.A.; Laarakkers, C.; Poorthuis, B.J. Dimethylmethylene Blue-Based Spectrophotometry of Glycosaminoglycans in Untreated Urine: A Rapid Screening Procedure for Mucopolysaccharidoses. Clin. Chem 1989, 35, 1472–1477.
- Tomatsu, S.; Fujii, T.; Fukushi, M.; Oguma, T.; Shimada, T.; Maeda, M.; Kida, K.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; et al. Newborn Screening and Diagnosis of Mucopolysaccharidoses. Mol. Genet. Metab. 2013, 110, 42–53.
- Kakkis, E.; Marsden, D. Urinary Glycosaminoglycans as a Potential Biomarker for Evaluating Treatment Efficacy in Subjects with Mucopolysaccharidoses. Mol. Genet. Metab. 2020, 130, 7–15.
- Denamur, S.; Touati, G.; Debelleix, S.; Damaj, L.; Barth, M.; Tardieu, M.; Gorce, M.; Broué, P.; Lacombe, D.; Labarthe, F. Recommended Respiratory Tests Are Not Routinely Performed for Mucopolysaccharidosis Patients. ERJ Open Res. 2022, 8, 00567–2021.
- Sawamoto, K.; Stapleton, M.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments. Drugs 2019, 79, 1103–1134.
- Haskins, M.E. Animal Models for Mucopolysaccharidosis Disorders and Their Clinical Relevance. Acta Paediatr. 2007, 96, 56–62.
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246.
- Poe, M.D.; Chagnon, S.L.; Escolar, M.L. Early Treatment Is Associated with Improved Cognition in Hurler Syndrome. Ann. Neurol. 2014, 76, 747–753.
- Deduve, C. From Cytases to Lysosomes. Fed Proc. 1964, 23, 1045–1049.
- Jones, S.A.; Breen, C.; Heap, F.; Rust, S.; de Ruijter, J.; Tump, E.; Marchal, J.P.; Pan, L.; Qiu, Y.; Chung, J.-K.; et al. A Phase 1/2 Study of Intrathecal Heparan-N-Sulfatase in Patients with Mucopolysaccharidosis IIIA. Mol. Genet. Metab. 2016, 118, 198–205.
- Kan, S.-H.; Aoyagi-Scharber, M.; Le, S.Q.; Vincelette, J.; Ohmi, K.; Bullens, S.; Wendt, D.J.; Christianson, T.M.; Tiger, P.M.N.; Brown, J.R.; et al. Delivery of an Enzyme-IGFII Fusion Protein to the Mouse Brain Is Therapeutic for Mucopolysaccharidosis Type IIIB. Proc. Natl. Acad. Sci. USA 2014, 111, 14870–14875.
- Boado, R.J.; Lu, J.Z.; Hui, E.K.-W.; Lin, H.; Pardridge, W.M. Insulin Receptor Antibody-α-N-Acetylglucosaminidase Fusion Protein Penetrates the Primate Blood-Brain Barrier and Reduces Glycosoaminoglycans in Sanfilippo Type B Fibroblasts. Mol. Pharm. 2016, 13, 1385–1392.
- Giugliani, R.; Dalla Corte, A.; Poswar, F.; Vanzella, C.; Horovitz, D.; Riegel, M.; Baldo, G.; Vairo, F. Intrathecal/Intracerebroventricular Enzyme Replacement Therapy for the Mucopolysaccharidoses: Efficacy, Safety, and Prospects. Expert Opin. Orphan Drugs 2018, 6, 403–411.
- Baldo, G.; Giugliani, R.; Matte, U. Gene Delivery Strategies for the Treatment of Mucopolysaccharidoses. Expert Opin. Drug Deliv. 2014, 11, 449–459.
- Sawamoto, K.; Chen, H.-H.; Alméciga-Díaz, C.J.; Mason, R.W.; Tomatsu, S. Gene Therapy for Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 123, 59–68.
- Wood, S.R.; Bigger, B.W. Delivering Gene Therapy for Mucopolysaccharide Diseases. Front. Mol. Biosci. 2022, 9, 965089.
- Tardieu, M.; Zérah, M.; Husson, B.; de Bournonville, S.; Deiva, K.; Adamsbaum, C.; Vincent, F.; Hocquemiller, M.; Broissand, C.; Furlan, V.; et al. Intracerebral Administration of Adeno-Associated Viral Vector Serotype Rh.10 Carrying Human SGSH and SUMF1 CDNAs in Children with Mucopolysaccharidosis Type IIIA Disease: Results of a Phase I/II Trial. Hum. Gene Ther. 2014, 25, 506–516.
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-Associated Virus Vector as a Platform for Gene Therapy Delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378.
- Eliyahu, E.; Wolfson, T.; Ge, Y.; Jepsen, K.J.; Schuchman, E.H.; Simonaro, C.M. Anti-TNF-Alpha Therapy Enhances the Effects of Enzyme Replacement Therapy in Rats with Mucopolysaccharidosis Type VI. PLoS ONE 2011, 6, e22447.
- Schuchman, E.H.; Ge, Y.; Lai, A.; Borisov, Y.; Faillace, M.; Eliyahu, E.; He, X.; Iatridis, J.; Vlassara, H.; Striker, G.; et al. Pentosan Polysulfate: A Novel Therapy for the Mucopolysaccharidoses. PLoS ONE 2013, 8, e54459.
- Guo, N.; DeAngelis, V.; Zhu, C.; Schuchman, E.H.; Simonaro, C.M. Pentosan Polysulfate Treatment of Mucopolysaccharidosis Type IIIA Mice. JIMD Rep. 2018, 43, 37–52.
- Kingma, S.D.K.; Wagemans, T.; IJlst, L.; Seppen, J.; Gijbels, M.J.J.; Wijburg, F.A.; van Vlies, N. Adverse Effects of Genistein in a Mucopolysaccharidosis Type I Mouse Model. JIMD Rep. 2015, 23, 77–83.
- Ghosh, A.; Rust, S.; Langford-Smith, K.; Weisberg, D.; Canal, M.; Breen, C.; Hepburn, M.; Tylee, K.; Vaz, F.M.; Vail, A.; et al. High Dose Genistein in Sanfilippo Syndrome: A Randomised Controlled Trial. J. Inherit. Metab. Dis. 2021, 44, 1248–1262.
- Losada Díaz, J.C.; Cepeda del Castillo, J.; Rodriguez-López, E.A.; Alméciga-Díaz, C.J. Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 232.
- Lindahl, U.; Couchman, J.; Kimata, K.; Esko, J.D. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015.
- Taylor, K.R.; Gallo, R.L. Glycosaminoglycans and Their Proteoglycans: Host-Associated Molecular Patterns for Initiation and Modulation of Inflammation. FASEB J. 2006, 20, 9–22.
- Sasisekharan, R.; Venkataraman, G. Heparin and Heparan Sulfate: Biosynthesis, Structure and Function. Curr. Opin. Chem. Biol. 2000, 4, 626–631.
- Xu, D.; Esko, J.D. Demystifying Heparan Sulfate-Protein Interactions. Annu. Rev. Biochem. 2014, 83, 129–157.
- Gandhi, N.S.; Mancera, R.L. The Structure of Glycosaminoglycans and Their Interactions with Proteins. Chem. Biol. Drug Des. 2008, 72, 455–482.
- Ly, M.; Leach, F.E.; Laremore, T.N.; Toida, T.; Amster, I.J.; Linhardt, R.J. The Proteoglycan Bikunin Has a Defined Sequence. Nat. Chem. Biol. 2011, 7, 827–833.
- Iozzo, R.V.; Schaefer, L. Proteoglycan Form and Function: A Comprehensive Nomenclature of Proteoglycans. Matrix Biol. 2015, 42, 11–55.
- Malmström, A.; Bartolini, B.; Thelin, M.A.; Pacheco, B.; Maccarana, M. Iduronic Acid in Chondroitin/Dermatan Sulfate: Biosynthesis and Biological Function. J. Histochem. Cytochem. 2012, 60, 916–925.
- Yamada, S.; Sugahara, K. Potential Therapeutic Application of Chondroitin Sulfate/Dermatan Sulfate. Curr. Drug Discov. Technol. 2008, 5, 289–301.
- Ambrosius, M.; Kleesiek, K.; Götting, C. Quantitative Determination and Comparison of the Glycosaminoglycan Delta-Disaccharide Composition in 22 Different Human Cell Lines. Cell Biol. Int. 2009, 33, 848–852.
- Malmström, A. Biosynthesis of Dermatan Sulfate. II. Substrate Specificity of the C-5 Uronosyl Epimerase. J. Biol. Chem. 1984, 259, 161–165.
- Mizumoto, S.; Sugahara, K. Glycosaminoglycan Chain Analysis and Characterization (Glycosylation/Epimerization). Methods Mol. Biol. 2012, 836, 99–115.
- Thelin, M.; Svensson, K.J.; Shi, X.; Bagher, M.; Axelsson, J.; Isinger-Ekstrand, A.; van Kuppevelt, T.H.; Johansson, J.; Nilbert, M.; Zaia, J.; et al. Dermatan Sulfate Is Involved in the Tumorigenic Properties of Esophagus Squamous Cell Carcinoma. Cancer Res. 2012, 72, 1943–1952.
- Morimoto, H.; Kida, S.; Yoden, E.; Kinoshita, M.; Tanaka, N.; Yamamoto, R.; Koshimura, Y.; Takagi, H.; Takahashi, K.; Hirato, T.; et al. Clearance of Heparan Sulfate in the Brain Prevents Neurodegeneration and Neurocognitive Impairment in MPS II Mice. Mol. Ther. 2021, 29, 1853–1861.
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of Mucopolysaccharidoses, an Update. Int. J. Mol. Sci. 2020, 21, 2515.
- Minami, K.; Morimoto, H.; Morioka, H.; Imakiire, A.; Kinoshita, M.; Yamamoto, R.; Hirato, T.; Sonoda, H. Pathogenic Roles of Heparan Sulfate and Its Use as a Biomarker in Mucopolysaccharidoses. Int. J. Mol. Sci. 2022, 23, 11724.
- De Pasquale, V.; Pavone, L.M. Heparan Sulfate Proteoglycans: The Sweet Side of Development Turns Sour in Mucopolysaccharidoses. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165539.
- Berger, K.I.; Fagondes, S.C.; Giugliani, R.; Hardy, K.A.; Lee, K.S.; McArdle, C.; Scarpa, M.; Tobin, M.J.; Ward, S.A.; Rapoport, D.M. Respiratory and Sleep Disorders in Mucopolysaccharidosis. J. Inherit. Metab. Dis. 2013, 36, 201–210.
- Chazeirat, T.; Denamur, S.; Bojarski, K.K.; Andrault, P.-M.; Sizaret, D.; Zhang, F.; Saidi, A.; Tardieu, M.; Linhardt, R.J.; Labarthe, F.; et al. The Abnormal Accumulation of Heparan Sulfate in Patients with Mucopolysaccharidosis Prevents the Elastolytic Activity of Cathepsin V. Carbohydr. Polym. 2021, 253, 117261.
- Sandri, G.; Bonferoni, M.C.; Rossi, S.; Delfino, A.; Riva, F.; Icaro Cornaglia, A.; Marrubini, G.; Musitelli, G.; Del Fante, C.; Perotti, C.; et al. Platelet Lysate and Chondroitin Sulfate Loaded Contact Lenses to Heal Corneal Lesions. Int J. Pharm. 2016, 509, 188–196.
- Ronca, F.; Palmieri, L.; Panicucci, P.; Ronca, G. Anti-Inflammatory Activity of Chondroitin Sulfate. Osteoarthr. Cartil. 1998, 6 (Suppl. A), 14–21.
- Kim, J.S.; Werth, V.P. Identification of Specific Chondroitin Sulfate Species in Cutaneous Autoimmune Disease. J. Histochem. Cytochem. 2011, 59, 780–790.
- Kai, Y.; Yoneyama, H.; Yoshikawa, M.; Kimura, H.; Muro, S. Chondroitin Sulfate in Tissue Remodeling: Therapeutic Implications for Pulmonary Fibrosis. Respir. Investig. 2021, 59, 576–588.
- Trowbridge, J.M.; Gallo, R.L. Dermatan Sulfate: New Functions from an Old Glycosaminoglycan. Glycobiology 2002, 12, 117R–125R.
- Zhang, B.; Chi, L. Chondroitin Sulfate/Dermatan Sulfate-Protein Interactions and Their Biological Functions in Human Diseases: Implications and Analytical Tools. Front. Cell Dev. Biol. 2021, 9, 693563.
- Harmatz, P.; Shediac, R. Mucopolysaccharidosis VI: Pathophysiology, Diagnosis and Treatment. Front. Biosci. (Landmark Ed.) 2017, 22, 385–406.
- Mucopolysaccharidosis Type VII: MedlinePlus Genetics. Available online: https://medlineplus.gov/genetics/condition/mucopolysaccharidosis-type-vii/ (accessed on 27 January 2023).
- Caterson, B.; Melrose, J. Keratan Sulfate, a Complex Glycosaminoglycan with Unique Functional Capability. Glycobiology 2018, 28, 182–206.
- Pomin, V.H. Keratan Sulfate: An up-to-Date Review. Int. J. Biol. Macromol. 2015, 72, 282–289.
- Quantock, A.J.; Young, R.D.; Akama, T.O. Structural and Biochemical Aspects of Keratan Sulphate in the Cornea. Cell Mol. Life Sci. 2010, 67, 891–906.
- Conrad, A.H.; Zhang, Y.; Tasheva, E.S.; Conrad, G.W. Proteomic Analysis of Potential Keratan Sulfate, Chondroitin Sulfate A, and Hyaluronic Acid Molecular Interactions. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4500–4515.
- Tomatsu, S.; Gutierrez, M.; Nishioka, T.; Yamada, M.; Yamada, M.; Tosaka, Y.; Grubb, J.H.; Montaño, A.M.; Vieira, M.B.; Trandafirescu, G.G.; et al. Development of MPS IVA Mouse (Galnstm(HC79S.MC76S)Slu) Tolerant to Human N-Acetylgalactosamine-6-Sulfate Sulfatase. Hum. Mol. Genet. 2005, 14, 3321–3335.
- Dũng, V.C.; Tomatsu, S.; Montaño, A.M.; Gottesman, G.; Bober, M.B.; Mackenzie, W.; Maeda, M.; Mitchell, G.A.; Suzuki, Y.; Orii, T. Mucopolysaccharidosis IVA: Correlation between Genotype, Phenotype and Keratan Sulfate Levels. Mol. Genet. Metab. 2013, 110, 129–138.
- Tomatsu, S.; Okamura, K.; Taketani, T.; Orii, K.O.; Nishioka, T.; Gutierrez, M.A.; Velez-Castrillon, S.; Fachel, A.A.; Grubb, J.H.; Cooper, A.; et al. Development and Testing of New Screening Method for Keratan Sulfate in Mucopolysaccharidosis IVA. Pediatr. Res. 2004, 55, 592–597.
- Vallet, S.D.; Clerc, O.; Ricard-Blum, S. Glycosaminoglycan-Protein Interactions: The First Draft of the Glycosaminoglycan Interactome. J. Histochem. Cytochem. 2021, 69, 93–104.
- Leal, A.F.; Benincore-Flórez, E.; Rintz, E.; Herreño-Pachón, A.M.; Celik, B.; Ago, Y.; Alméciga-Díaz, C.J.; Tomatsu, S. Mucopolysaccharidoses: Cellular Consequences of Glycosaminoglycans Accumulation and Potential Targets. Int. J. Mol. Sci. 2022, 24, 477.
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine Cathepsins: From Structure, Function and Regulation to New Frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88.
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine Cathepsins and Their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264.
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679.
- Lecaille, F.; Chazeirat, T.; Saidi, A.; Lalmanach, G. Cathepsin V: Molecular Characteristics and Significance in Health and Disease. Mol. Aspects Med. 2022, 88, 101086.
This entry is offline, you can click here to edit this entry!