Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Natural products and plant extracts exhibit many biological activities, including that related to the defense mechanisms against parasites. The pandemic emergencies have further increased the interest in finding antiviral agents, and efforts are oriented to investigate possible activities of secondary plant metabolites against human viruses and their potential application in treating or preventing SARS-CoV-2 infection.
- phytochemicals
- secondary metabolites
- antiviral activity
- SARS-CoV-2
- COVID-19
1. Introduction
Plants produce a variety of structurally diverse and often complex secondary metabolites (SM) with a range of biological functions [1]. Plant SM can be classified according to several criteria, such as chemical structure (presence of rings or sugars), composition (containing nitrogen or not), solubility in organic solvents or water, and the biosynthetic pathway [2]. They include alkaloids, carotenoids, organosulfur compounds, phytosterols, nitrogen compounds, and phenolics (Figure 1).
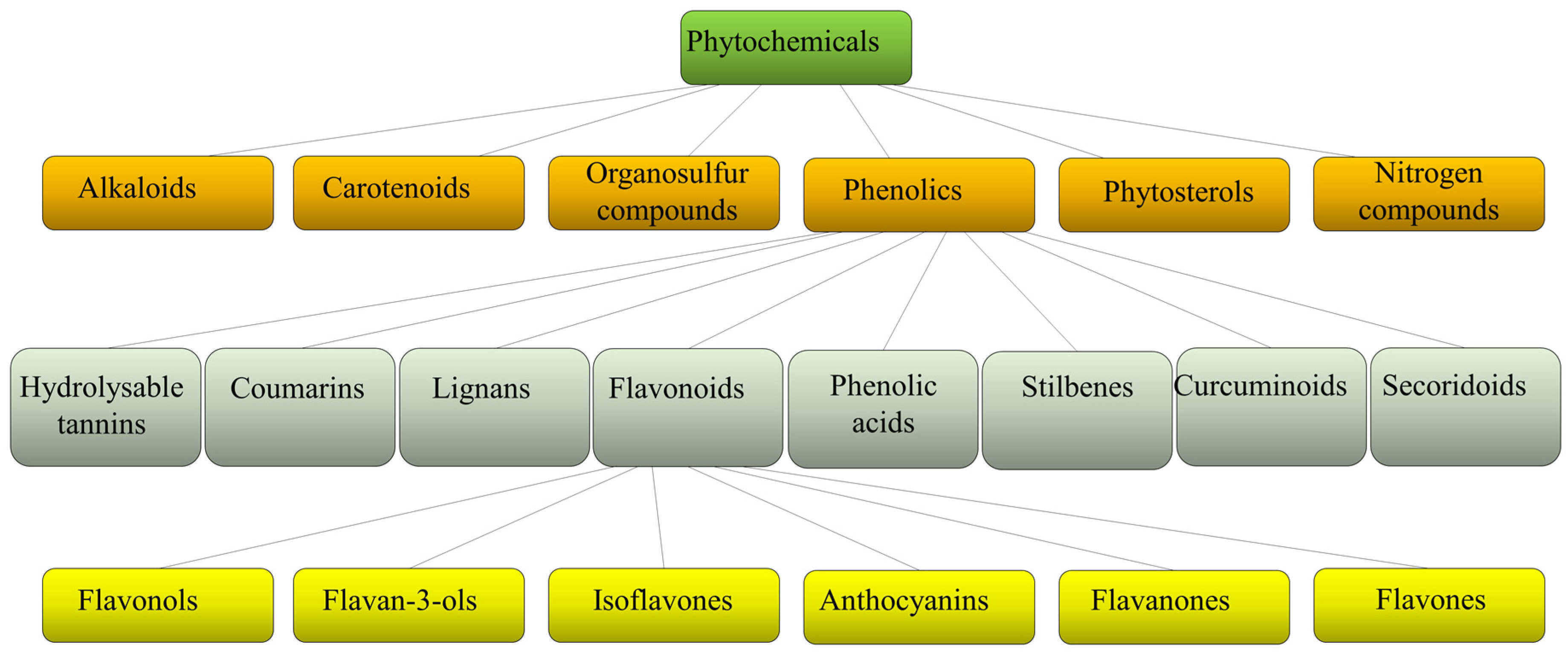
Figure 1. Schematic classification of phytochemicals, with sub-classification of phenolics and flavonoids.
Each class is then divided into further classes. In particular, phenolics comprise a large group of different compounds, with the phenolic hydroxyl groups being the common structural feature. These are usually found conjugated with sugars and organic acids and can be divided into seven main classes such as hydrolyzable tannins, coumarins, lignans, phenolic acids, stilbenes, curcuminoids, and flavonoids [3] (Figure 1). Flavonoids exist broadly in nature, and to date, more than 9000 flavonoids have been reported. They can be divided into the following subclasses: flavonols, flavan-3-ols, isoflavones, anthocyanins, flavanones, and flavones. Flavonoids share a common structure of two benzene rings connected by three carbon atoms forming an oxygenated heterocycle and are responsible for the red, blue, and yellow coloration of plants and are found in foods and beverages of plant origin, such as fruits, vegetables, tea, cocoa, and wine [4][5]. Recent studies have reported that some of these natural compounds can reduce the risk of many chronic diseases and can significantly modulate and diversify the composition of the human gut microbiome [6]. Consumption of phenolic compounds can help to prevent metabolic, cardiovascular, respiratory, neurological, and cancerous diseases [7][8] due to their high antimicrobial, antioxidant, and anti-inflammatory immunomodulatory activities [9][10][11]. Eating plant-based foods is part of many diverse dietary patterns, including the well-studied Mediterranean diet [12], vegan, and vegetarian approaches. Despite the widely known health benefits of consuming fruits and vegetables, the intake is often inadequate and a large number of adults worldwide do not consume the daily-recommended servings. So, in the attempt to improve health, dietary guidelines and health promotion campaigns have advocated for individuals to “eat a rainbow” of fruit and vegetables, i.e., to take a qualitative color rather than a quantitative servings approach, based on the association of each color with a health benefit. For example, red foods include antioxidants that can contribute to decreased inflammation in the body; orange foods are abundant in carotenoids and have been linked to endocrine-regulating activities; yellow foods have been found to aid in digestion due to their fiber content and bioflavonoids that promote healthy gut bacteria; green foods, especially green leafy vegetables, contain an abundance of polyphenols that aid in reducing cardiovascular risk factors such as high blood pressure; blue or purple foods have been found to improve memory and mood because of flavonoids, flavonols, and phenolic acids that promote cognitive functions.
Recent technological advances in analytical methods such as metabolomics, metabolic engineering, and synthetic biology, as well as the ad hoc designed computational tools and databases, are providing powerful tools for drug discovery based on natural compounds [6]. It has been known that natural products and plant extracts exhibit potent anti-viral activities, often against multiple virus families, thus suggesting that they may be useful as broad-spectrum antiviral agents [13][14].
The COVID-19 pandemic caused by SARS-CoV-2 has prompted researchers to conduct many studies aimed at identifying useful compounds to counter the viral agent. As reported by the World Health Organization’s weekly epidemiological update dated 22 February 2023, “over 757 million confirmed cases and over 6.8 million deaths have been reported globally” [15], with nearly 5.3 million new cases and over 48,000 deaths in the 4 weeks preceding the report. For a state-of-the-art review of the pandemic two years after its appearance and the related lessons learned, an interesting article was published in September 2022 [16].
2. Antiviral Potential of Selected Natural Phytochemicals
Natural products serve as excellent sources for discovering antiviral agents due to their diversity and complexity and can offer remarkable efficacy and specificity to target viral infections [17]. The antiviral activity of a drug can affect viral entry, viral DNA and RNA synthesis, or viral reproduction, and is conditioned by viral structure and its replication cycle. For example, the presence or absence of a viral envelope, and, consequently, the different modes of entry of the virus into the host cell, play a significant role in the effectiveness of the virucidal activity, as the surface of the external membrane of the virus establishes the first contact with the drug. The antiviral activity of secondary metabolites can be evaluated by various biological assays to test the cytotoxicity, cytopathic effect inhibition, and the capability to block viral spread from cell to cell, thus limiting and/or fighting viral circulation [18]. Several natural products exhibit antiviral activities towards DNA and RNA viruses by acting on different cellular/viral targets and interfering with the infection and the viral replication cycle. Many of the secondary metabolites that have shown antiviral activity against the Herpes simplex virus (HSV). HSV belongs to Herpesviridae, which is a broad family of enveloped-DNA viruses and is categorized into two types: HSV-1, commonly associated with orofacial ulcers, and HSV-2, which mainly causes genital ulcers [19]. For example, caffeic acid inhibits Herpes simplex type 1 (HSV-1) multiplication mainly before the completion of viral DNA replication but not thereafter [20]. Gallic acid, also belonging to the class of phenolic acids, has shown activity against HSV [21]. In the context of flavonoids, luteolin—belonging to the class of flavones—exhibited potent antiviral activity against wild-type and clinical isolates of HSV-2 [22], as well as kaempferol and its glycosides kaempferol-3-O-robinobioside and kaempferol-3-O-rutinoside, myricetin, quercetin, and its glycosides quercetin-3-O-rutinoside (rutin), and isorhamnetin, all belonging to the class of flavonols that have shown inhibitory activity against HSV-1 and/or HSV-2 [21][23][24][25][26]. The flavanols epicatechin, (-)-epicatechin-3-O-gallate (ECG), (-)-epigallocatechin (EGC), and epigallocatechin-3-gallate (EGCG) and the flavanone naringenin possess antiherpetic activity [21][27]. In vitro studies have also been performed on molecules belonging to other classes of phenols such as curcumin [19], resveratrol [28], chebulagic acid [29], and other secondary metabolites such as allicin [30] and eugenol [31], demonstrating their antiviral activity against HSV-1 and HSV-2.
Furthermore, studies have investigated the antiviral activity of some secondary metabolites against the Hepatitis C virus (HCV), a virus belonging to the Flaviviridae family that represents one of the major causes of chronic liver disease. It has been demonstrated that caffeic acid, as well as the flavone apigenin, could inhibit HCV replication [23][32], while gallic acid, quercetin-3-O-rutinoside (rutin), epigallocatechin-3-gallate (EGCG), and curcumin could inhibit HCV entry [19][27][33][34]. In hepatitis virus infection, coumarin has been shown to target a wide range of proteins, like binding antigens present at the cell surface, proteins involved in viral replication, and interferon signaling pathways [35]. Furthermore, it has been demonstrated that sulforaphane, an isothiocyanate widely distributed in Brassicaceae plants, suppresses the replication of HCV by inducing the heme-oxygenase-1 (HO-1) expression, an enzyme that interferes with the replication of the virus through the activation of nuclear factor (erythroid-derived 2)-like (Nrf2) pathway, which regulates the expression of antioxidant proteins [36].
Adenoviruses (ADVs) are DNA viruses that typically cause mild infections involving the upper or lower respiratory tract, gastrointestinal tract, or conjunctiva. More than 50 serotypes have been identified showing different tissue tropisms that correlate with clinical manifestations of infection [37]. In a study conducted to examine the antiviral activity of aqueous extract and pure compounds of Plantago major L., a popular traditional Chinese medicine, it was found that chlorogenic acid was active against ADV-3, ADV-8, and ADV-11; instead, caffeic acid was active against ADV-3 [38]. Epigallocatechin-3-gallate (EGCG) has been shown to reduce the virus titers of adenovirus in two cell infection models, to inactivate purified adenovirions and to inhibit the attachment of adenovirus by interacting with virion surface proteins [27]. Caffeic acid also inhibits the multiplication of the Influenza A (IAV) virus in the early stage of viral multiplication [39]. Influenza virus (IV) is a single-stranded RNA virus, which belongs to the Orthomyxoviridae family, that causes acute respiratory infection. There are four types of influenza viruses: A, B, C, and D. Human influenza A and B viruses cause seasonal epidemics of disease; influenza C virus infections generally cause mild illness and are not thought to cause human epidemics; influenza D viruses primarily affect cattle and are not known to infect or cause human illness. Type A virus is divided into subtypes according to the antigenic properties of surface glycoproteins, hemagglutinin (HA), and neuraminidase (NA). In relation to genetic and antigenic variation, the current 18 subtypes of IAV HA (H1–H18) are divided into group 1 (H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, H17, and H18) and group 2 (H3, H4, H7, H10, H14, and H15), while IAV of NA has 11 subtypes (N1–N11) [40]. Gallic acid has been shown to exhibit inhibitory effects against both influenza type A and B viruses, disrupting the viral particles [41], while hydroxytyrosol inactivates influenza A viruses, including H1N1, H3N2, H5N1, and H9N2 subtypes with an effect virucidal rather than antiviral [42]. Apigenin, luteolin, kaempferol, epicatechin, (-)-epicatechin-3-O-gallate (ECG), epigallocatechin-3-gallate (EGCG), cirsimaritin, and eugenol also show activity against IV inhibiting replication [23][24][27][43][44]. An in vitro and in vivo study demonstrated that isorhamnetin exerts anti-influenza effects via direct HA and NA inhibition, direct or indirect inhibition of the expression of viral HA and NA genes as well as reduces virus-induced ROS (reactive oxygen species) generation [45]. Several in vitro studies demonstrated that curcumin inhibits the uptake, replication, and particle production of different IAVs [19]. In an in vitro and in silico study, Sang et al. [46] demonstrated that myricetin has not only a direct antiviral activity against IAV-inhibiting virus replication but also has immune modulatory effects. Wu et al. [47] demonstrated that quercetin inhibits a wide spectrum of influenza groups, including H1N1, H3N2, and H5N1, binding to influenza hemagglutinin protein and inhibiting viral-cell fusion.
Instead, quercetin 3-rhamnoside (quercitrin) has a strong antiviral activity against the influenza A/WS/33 (H1N1) virus by inhibiting replication in the initial phase of the infection by indirect interaction with viral particles [48]. Sahoo et al. [49] used in silico docking approaches to explore the molecular interaction of 13 active compounds extracted from plants against NA protein of H1N1 and observed that theaflavin, found in green tea, could be a potential inhibitor of H1N1 NA proteins, as strongly suggested by lowest docking energy. Molecular docking studies have also shown the inhibitory role against IV infection of coumarin and its derivatives that can target various enzymes and pathways essential for viral entry, survival, and infection [35]. An in vitro study on the effect of sulforaphane on IAV replication in Madin-Darby canine kidney cells showed a decrease in replication [36].
The Human immunodeficiency virus (HIV), a human retrovirus belonging to the lentivirus family that attacks the body’s immune system, is responsible for causing acquired immunodeficiency syndrome (AIDS) once the immune system is compromised. There are two known types of HIV, which are deeply related, named HIV-1 and HIV-2. HIV-1 is the main cause of AIDS and responsible for the worldwide HIV pandemic; HIV-2, which differs in genomic structure and antigenicity, also causes the disease but with slower progression [50]. Different plants and phytochemicals exert activity against HIV. For example, gallic acid and ellagic acid possess anti-HIV activity and may act as post-entry inhibitors by affecting HIV protease activity [51]. Hydroxytyrosol also shows anti-HIV activity in vitro, inhibiting the viral integrase enzyme and fusion of the viral envelope with host cells [52]. Apigenin, as well as epicatechin, (-)-epicatechin-3-O-gallate (ECG), and epigallocatechin-3-gallate (EGCG), exhibit antiviral activities against HIV, presumably inhibiting HIV-1 protease enzyme and HIV reverse transcriptase [23][27]. Luteolin inhibits HIV-1 at the transcription level after the viral integration step [24], while myricetin inhibits the HIV-1 integrase (IN) and the HIV-1 reverse transcriptase [53]. The results from Ortega and co-workers [54] showed that the differences in the antiviral activity and the RT inhibition of the myricetin and myricetin-3-rhamnoside (myricitrin) together with the in silico analysis suggested that the glycosyl moiety could play a role in the entry of flavonoids into the cell and then, after enzymatic cleavage of the glycosyl moiety, the myricetin aglycone would ultimately be responsible for the anti-HIV activity. Quercetin has been investigated in vitro as an antiviral agent for HIV due to its ability to inhibit crucial enzymes such as reverse transcriptase, integrase, and protease [55]. Resveratrol also has effects on viral infection by inhibiting HIV-1 strain replication [24]. Several studies have reported that curcumin exhibits anti-HIV activity by directly targeting viral proteins. In particular, curcumin shows inhibition of the HIV-1 and HIV-2 proteases, the HIV integrase and the HIV trans-activator of transcription (tat), a viral transcription regulator [19]. Coumarin derivatives also exhibit their anti-HIV activity by targeting viral proteins inhibiting HIV protease, integrase, reverse transcriptase, and tat but also by inhibiting viral DNA replication [35]. Sulforaphane protects macrophages from HIV infection by mobilizing the transcription factor and antioxidant response regulator nuclear factor E2-related factor 2 (Nrf2) [36].
Enterovirus 71 (EV71), which belongs to the genus Enterovirus, is the major pathogen of hand, foot, and mouth disease (HFMD) that often occurs in children under the age of 10 [56]. Gallic acid was found to exert strong anti-EV71 activity [56], while isorhamnetin inhibits EV71 RNA replication and protein synthesis [23]. It has been demonstrated that resveratrol inhibits EV71 replication [57]. Antiviral activity of epigallocatechin-3-gallate (EGCG) and coumarin and its derivatives has been observed against EV71 [27][35]. Chebulagic acid and Punicalagin exhibit antiviral activity both in vitro and in vivo against EV71 [58][59].
3. Natural Phytocompounds with Potential to Inhibit the Coronavirus SARS-CoV-2 According to In Silico Approaches
In silico molecular modeling uses the crystal structure of viral targets from SARS-CoV-2, or targets of related viruses such as SARS-CoV, docked with molecules under study to establish the binding strength of competitive inhibitors of the target that is expressed in terms of Kcal/mol. The binding strength of a test compound is compared with that of a known inhibitor or control. The in silico docking is no indication that a compound will absolutely inhibit a target, but it offers a good start for engaging in drug design [60]. Researchers have performed an analysis of studies carried out with an in silico approach of possible interactions of the same molecules analyzed above for their antiviral activity with the major targets of SARS-CoV-2, like the main protease (MPro), RdRp, membrane (M) glycoprotein, envelope (E) glycoprotein, spike (S) glycoprotein, PLpro, non-structural proteins (NSP) and ACE2 receptor. In particular, for those molecules that have shown activity against SARS-CoV, it has been postulated that they could potentially act against SARS-CoV-2, taking into account that SARS-CoV and SARS-CoV-2 have a high sequence similarity (79.5%) [61]. For example, Alrasheid and co-workers [62] have conducted an in silico study by using Molecular Operating Environment (MOE) drug discovery software platform in order to evaluate the antiviral activity targeting SARS-CoV-2 of twenty-one compounds from medicinal plants. They have found that gallic acid, quercetin, and capsaicin were among the best compounds interacting with SARS-CoV-2 Mpro, with rank scores ranging from −17.45 to −13.90 Kcal/mol. Moreover, a screening via molecular docking to test the binding affinity of various selected bioactive compounds of honey and propolis as inhibitors against the SARS-CoV-2 Mpro and RdRp revealed that ellagic acid and kaempferol have the strongest interaction with RdRp, instead the previously cited compounds and quercetin with the SARS-CoV-2 Mpro [63]. In another study, in which one hundred secondary metabolites from Aframomum melegueta, a Zingiberaceae family plant spice widely spread in Africa, have been computationally evaluated for inhibition of SARS-CoV-2 targets, it is reported that quercetin could be a potential SARS-CoV-2 2′-O-methyltransferase (NSP16) enzyme inhibitor while apigenin could inhibit SARS-CoV-2 main protease (Mpro) [64]. In a 2020 study published as a preprint, kaempferol, quercetin, naringenin, oleuropein, curcumin, catechin, and epicatechin-gallate appeared to have the best potential to act as SARS-CoV-2 Mpro inhibitors with binding energies values obtained from the docking of main protease of −8.58, −8.47, −7.89, −7.31, −7.05, −7.24, and −6.67 Kcal/mol, respectively [65]. Although the preprint was not published elsewhere to date, other studies confirm the results. Bilginer et al. [66] also showed similar data for kaempferol, quercetin, and oleuropein; instead, Halder et al. [67] for curcumin and catechin. Moreover, Mukheriee et al. [68] confirmed a possible binding of epicatechin-gallate, quercetin, and kaempferol to the Mpro active site, with binding energies values of −8.5, −7.6, and −7.3 Kcal/mol respectively, and underline as epigallocatechin-gallate shows the best affinity in comparison to the re-docked inhibitor binding energy (−7.7 Kcal/mol).
In another in silico study, published as a preprint, luteolin was also screened against SARS-CoV-2 main protease (Mpro), showing binding energy values of −8.35 Kcal/mol [35]; also, in this case, further studies corroborate this hypothesis [69]. Based on the inhibitory activity against SARS-CoV 3CLpro exerted by pectolinarin, rhoifolin, and herbacetin [70][71] performed an in silico docking study to deduce their binding mode and binding affinity with SARS-CoV-2 3CLpro and found that the affinity of rhoifolin became weakened while the efficiency of herbacetin and pectolinarin was still promising. Vicidomini et al. [72] found that for several flavonols and flavonol glucosides isolated from Opuntia ficus-indica, a dicotyledonous angiosperm cactaceous plant widespread worldwide in tropical and subtropical regions, including isorhamnetin, possible affinities for SARS-CoV-2 Mpro, with binding energies lower than −7.0 Kcal/mol. Myricetin-3-rhamnoside (myricitrin) was found to have a strong binding with the active site residues of the SARS-CoV-2 Mpro with a binding score of −8.9 Kcal/mol. In particular, its active site residues Tyr54, Phe140, Gly143, His163, and Glu166 are involved in the formation of 6 hydrogen bonds with hydroxyl and carboxyl oxygen of ligand while His41 and Cys145 form Pi-alkyl and Pi-sulfur interactions with the aromatic scaffold of ligand, respectively. Other additional interactions, such as van der Waals forces, stabilize the binding of the ligand to the enzyme [73]. In a study in which thirty-eight flavonoids have been tested by molecular docking against the active site of the SARS-CoV-2 Mpro, it has been demonstrated that natural aglycone flavonoids possess higher docking energies than flavonoids with sugar moieties. As a matter of fact, in these docking experiments, the aglycone myricetin displays a binding score of −7.4 Kcal/mol while myricetin-3-rhamnoside (myricitrin), also in this study, displays a binding score of −8.9 Kcal/mol. Similarly, quercetin-3-O-rhamnoside (quercitrin), quercetin-3-O-rutinoside (Rutin), quercetin-3-beta-galactoside, and quercetin 3-O-β-glucuronide exhibit lower binding energies than their aglycone quercetin (binding score of −9.7, −9.2, −8.4, −8.1, −7.5 Kcal/mol, respectively) [74]. Catechins and some of their derivatives showed a strong affinity for SARS-CoV-2 Mpro because they could form a stable ligand-receptor complex, and this leads to the hypothesis of a potential antiviral activity [75]. An in silico analysis has depicted that ellagic acid, epicatechin, and capsaicin exhibit significant binding and interaction with the most vital active site residue Cys145 of SARS-CoV-2 Mpro and can inhibit its activity in an extremely effective manner [76]. Ghosh et al. [77] selected eight polyphenols from green tea and elucidated the binding affinities and binding modes between these polyphenols and SARS-CoV-2 Mpro using molecular docking studies. All eight polyphenols exhibit good binding affinity, including catechin and (-)-epigallocatechin (EGC), but epigallocatechin-3-gallate (EGCG) and (-)-epicatechin-3-O-gallate (ECG) interact strongly with one or both the catalytic residues His41 and Cys145 of SARS-CoV-2 Mpro with a binding score of −7.6 and −8.2 Kcal/mol. Another study aimed to evaluate bioactive compounds found in plants to interact and hopefully inhibit SARS-CoV-2 Mpro and spike (S) glycoprotein using a molecular docking approach reported that pectolinarin, epigallocatechin-3-gallate (EGCG) and rhoifolin are among potential candidates to bind spike (S) glycoprotein surface, while rhoifolin and pectolinarin are among the candidates to become drugs targeting SARS-CoV-2 Mpro [78]. The role of tea polyphenols in prophylaxis and treatment of COVID-19 has been investigated by Mhatre et al. [79] in a preliminary in silico study in which molecular docking interactions of two tea polyphenols with some of the possible binding sites of SARS-CoV-2 were performed. In particular, the receptors 3CLpro, RdRp, PLpro, spike (S) glycoprotein RBD, and ACE2 receptor with spike (S) glycoprotein RBD were docked against epigallocatechin-3-gallate (EGCG) from green tea. For all the receptors studied, EGCG exhibited good docking scores with various types of interactions, and except for RdRp, the binding energies were consistently better than −8.0 Kcal/mol. Mostafa et al. [80] have conducted an in silico molecular docking experiment on the effects of some natural antiviral phytoconstituents on the crystal structure of SARS-CoV-2 Mpro. Many of the docked compounds revealed good binding affinity, with theaflavin showing a good binding score (−8.5 Kcal/mol). The bioactive phenolic phytocompound 6-gingerol, found in the fresh ginger rhizome, was also tested against different viral proteins by using the molecular docking technique in order to evaluate its interaction with SARS-CoV-2 targets. This study suggests a high binding affinity and interaction with surface regions of multiple targets of COVID-19, including viral proteases, RdRp, and spike (S) glycoprotein [81]. Du et al. [82] investigated in vitro and in silico the potent antiviral activity of chebulagic acid and punicalagin against SARS-CoV-2 viral replication. In silico docking of chebulagic acid and punicalagin to SARS-CoV-2 Mpro was performed in order to search potential allosteric binding sites, and both chebulagic acid and punicalagin may interact with the cleft between domain II and domain III within SARS-CoV-2 Mpro with stable binding free energy.
It is reported that SARS-CoV-2 can bind to Toll-like receptor 4 (TLR-4), which would eventually lead to α-synuclein aggregation in neurons and stimulation of neurodegeneration pathways. Oleuropein was investigated against the SARS-CoV-2 target (main protease 3CLpro), TLR-4, and Prolyl Oligopeptidases (POP), to explore oleuropein potency against the neurological complications associated with COVID-19. POP inhibition could reduce aggregation of alpha-synuclein in neurons, hence, investigating anti-COVID agent(s) against TLR-4 and POP might help in alleviating neurological complications linked with COVID-19. Docking analyses showed a binding score of −7.8, −8.3, and −8.5 Kcal/mol for oleuropein-3CLpro, oleuropein-TLR4, and oleuropein-POP interactions, respectively, suggesting oleuropein as a potential candidate that can target SARS-CoV-2 and alleviate neurological manifestations associated with it [83]. In a recent study, the antiviral potential of 50 natural coumarin phytochemicals isolated from plants was investigated in order to identify the binding interactions of these phytochemicals against the coronavirus 3CLpro by molecular modeling approaches. It was found that glycycoumarin, Inophyllum P, mesuol, and oxypeucedanin hydrate displayed the highest binding affinity with the best negative energy scores and interacted with one or both of the catalytic residues (His41 and Cys145) of 3CLpro through hydrophilic and hydrophobic bonding [84]. Shekh et al. [85], using virtual screening methods, have analyzed allicin to assess its ability to covalently modify cysteine residues of SARS-CoV-2 Mpro. The results suggest that allicin may induce dual S-thioallylation of Cys145 and Cys85/Cys156 residues of SARS-CoV-2 Mpro and may be useful in attenuating the coronavirus infection. It is known that essential oils have anti-inflammatory, immunomodulatory, bronchodilatory, and antiviral properties and, for this reason, are being proposed to have activity against SARS-CoV-2 virus. Moreover, owing to their lipophilic nature, essential oils are advocated to penetrate viral membranes easily, leading to membrane disruption. Molecular docking techniques were applied to screen the anti-SARS-CoV-2 efficacies of eugenol, menthol, and carvacrol, major components of essential oils, against various protein targets of SARS-CoV-2. Results revealed that these compounds have binding affinities towards SARS-CoV-2 spike (S) glycoprotein, SARS-CoV-2 Mpro, RdRp, and human ACE2 proteins, respectively [31].
Paolacci and co-workers [86] investigated the potential effects of hydroxytyrosol in combination with alpha-cyclodextrin, another naturally occurring compound, on SARS-CoV-2 entry into human cells. Bioinformatic docking studies showed that hydroxytyrosol is captured in the hydrophobic cavity of alpha-cyclodextrin, and the resulting complex could interact with the SARS-CoV-2 spike (S) glycoprotein and its host cell receptor ACE2. Even if these bindings do not occur at the spike-ACE2 interface, they may induce structural perturbations that could potentially influence the endocytosis process. Sekiou et al. [87] have screened in silico the interaction between the main protease SARS-CoV-2 Mpro active site with natural compounds, including cirsimaritin and quercetin, and found that both have a better binding affinity to this target. Molecular docking was used by Pandey et al. [24] to describe the binding ability of naturally occurring phytochemicals, including kaempferol and quercetin, with SARS-CoV-2 spike (S) glycoprotein and found that these natural compounds are capable of binding to either the S1 or S2 domains of the SARS-CoV-2 S, most probably preventing it from binding to the ACE2 receptor or internalization during fusion.
High-end molecular docking analysis and MD simulation were performed to characterize the binding affinity of natural and synthetic anti-viral compounds with the SARS-CoV-2 structural proteins and to analyze the stability of drug-protein interactions. Results identified rutin as a potent inhibitor of SARS-CoV-2 envelope (E) glycoprotein, while caffeic acid and ferulic acid were found to inhibit SARS-CoV-2 membrane (M) glycoprotein [88]. Orfali et al. [89] screened all available SARS-CoV-2 molecular targets using a multistep in silico protocol to find out the most probable one that mediates the proved in vitro anti-SARS-CoV-2 activity of sinapic acid and found that the viral envelope protein (E-protein) was suggested as the most probable hit for sinapic acid. A further in-depth molecular dynamic simulation-based investigation was performed to precisely explore the binding path and mode of sinapic acid with E-protein.
The interactions of the two crucial proteins NSP9 and NSP10 of COVID-19 have been investigated with potential antiviral compounds from Moringa oleifera using molecular docking and dynamic methods. The results revealed that all selected ligands form stable complexes with the targeted proteins and showed the highest binding affinity values of apigenin (−7.1 Kcal/mol) for NSP10 and ellagic acid (−7.1 Kcal/mol) for NSP9 [90]. Several viral proteins have been found to possess deubiquitinating activity, and they can be used to antagonize or modulate the antiviral immune signaling pathway. Along with protease activity, SARS-CoV-2 PLpro possesses deubiquitinating activity. Naphthalene-based inhibitors, such as the well-investigated GRL-0617 compound, have been shown to possess dual effects, inhibiting both protease and deubiquitinating activity of the PLpro. Pitsillou et al. [91] investigated the binding characteristics of the same dietary compounds, including rutin and cyanidin-3-O-glucoside, to the PLpro and evaluated the deubiquitinating activity. Analyses of molecular docking highlighted the relatively high affinity of GRL-0617 and dietary compounds. A study was conducted on the most abundant pomegranate (Punica granatum) peel extract constituents, including punicalagin, with the aim of exploring their anti SARS-CoV-2 properties using in silico tools. The protein targets used in this study were: SARS-CoV-2 spike (S) glycoprotein, ACE2, furin, and TMPRSS2. Molecular docking results showed that all the analyzed ligands interacted through hydrogen bonds with amino acid residues at the S glycoprotein predicted druggable site, and punicalagin presented free binding energy of −7.312 Kcal/mol, with the ligand-protein complex stabilized through three hydrogen bonds (Asn343, Asn370, and Ser371). All tested pomegranate peel extract constituents showed significant binding affinity at the ACE2 predicted druggable site, with all ligand–protein complexes stabilized through hydrogen bonds, and punicalagin presented free binding energy of −7.144 Kcal/mol. Amino acid residue Lys441 was found to be important for the stabilization of punicalagin–ACE2 complex. Moreover, all pomegranate peel extract constituents formed stable complexes with furin, with punicalagin-free binding energy of −9.385 Kcal/mol. Punicalagin showed intensive interactions with TMPRSS2 amino acid residues at the predicted binding site with binding energy values of −7.358 Kcal/mol and with four hydrogen bonds. Polar interactions with Asn97 and Arg405 residues were essential for the stabilization of TMPRSS2 complexes with punicalagin [92]. Özdemir et al. [93] carried out molecular docking studies on five different proteins (Spike S1-subunit, NSP5, NSP12, NSP15, and NSP16) of the SARS-CoV-2 and two proteins (ACE2 and Vitamin K epoxide reductase complex subunit 1-VKORC1, a key enzyme in recycling reduced vitamin K, that plays an essential role in γ-carboxylation of vitamin K-dependent coagulation factors) of human and found that the best binding scores for 17 coumarins were determined for NSP12, with the highest score (−10.01 Kcal/mol) showed by 2-morpholinoethan-1-amine substituted coumarin.
In conclusion, it is necessary to emphasize that the potential strength of molecular simulations offers the possibility of carrying out both large-scale screening and targeted docking simulations quickly and with limited costs, but on the other hand, it is also necessary to apply rigorous procedures and carefully evaluate the results obtained. The COVID-19 pandemic has prompted many research groups to carry out docking simulations to provide useful information to the scientific community as soon as possible and to identify possible solutions to the emergency as quickly as possible. However, researchers found several studies published only in pre-print form, and therefore without having received an evaluation by experts; others were published in peer-reviewed journals but very few days after submission to the journal. Although peer-reviewed, the very short evaluation time makes the quality of the review at least questionable. The result is that in several literature studies, as either full articles or pre-prints, there are poor protocols of docking simulations, weak or erroneous interpretations of the results, use of protein structures that are not experimental results but obtained by insufficiently described modeling procedures, docking with portions of proteins without critical evaluation of the real utility. Here, we chose to quote only in silico simulation studies that meet sufficient quality criteria in the simulations performed and in the presentation and discussion of the results obtained.
This entry is adapted from the peer-reviewed paper 10.3390/molecules28062470
References
- Garagounis, C.; Delkis, N.; Papadopoulou, K.K. Unraveling the roles of plant specialized metabolites: Using synthetic biology to design molecular biosensors. New Phytol. 2021, 231, 1338–1352.
- Weremczuk-Jeżyna, I.; Hnatuszko-Konka, K.; Lebelt, L.; Grzegorczyk-Karolak, I. The Protective Function and Modification of Secondary Metabolite Accumulation in Response to Light Stress in Dracocephalum forrestii Shoots. Int. J. Mol. Sci. 2021, 22, 7965.
- Liu, R.H. Health bene fits of phytochemicals in whole foods. In Nutritional Health: Strategies for Disease Prevention, 3rd ed.; Temple, N.J., Wilson, T., Jacobs, D.R., Jr., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 293–310.
- Abou Baker, D.H. An ethnopharmacological review on the therapeutical properties of fla-vonoids and their mechanisms of actions: A comprehensive review based on up to date knowledge. Toxicol. Rep. 2022, 9, 445–469.
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47.
- Periwal, V.; Bassler, S.; Andrejev, S.; Gabrielli, N.; Patil, K.R.; Typas, A.; Patil, K.R. Bioactivity assessment of natural compounds using machine learning models trained on target similarity between drugs. PLoS Comput. Biol. 2022, 18, e1010029.
- Behl, T.; Rocchetti, G.; Chadha, S.; Zengin, G.; Bungau, S.; Kumar, A.; Mehta, V.; Uddin, M.S.; Khullar, G.; Setia, D.; et al. Phytochemicals from Plant Foods as Potential Source of Antiviral Agents: An Overview. Pharmaceuticals 2021, 14, 381.
- Tirado-Kulieva, V.; Atoche-Dioses, S.; Hernandez-Martínez, E. Phenolic compounds of mango (Mangifera indica) by-products: Antioxidant and antimicrobial potential, use in disease prevention and food industry, methods of extraction and microencapsulation. Sci. Agropecu. 2021, 12, 283–293.
- Tirado-Kulieva, V.A.; Gutierrez-Valverde, K.S.; Villegas-Yarleque, M.; Camacho-Orbegoso, E.W.; Villegas-Aguilar, G.F. Research trends on mango by-products: A literature review with bibliometric analysis. J. Food Meas. Charact. 2022, 16, 2760–2771.
- Pérez de la Lastra, J.M.; Andrés-Juan, C.; Plou, F.J.; Pérez-Lebeña, E. Impact of Zinc, Glutathione, and Polyphenols as Antioxidants in the Immune Response against SARS-CoV-2. Processes 2021, 9, 506.
- Mehany, T.; Khalifa, I.; Barakat, H.; Althwab, S.A.; Alharbi, Y.M.; El-Sohaimy, S. Polyphenols as promising biologically active substances for preventing SARS-CoV-2: A review with research evidence and underlying mechanisms. Food Biosci. 2021, 40, 100891.
- Davis, C.; Bryan, J.; Hodgson, J.; Murphy, K. Definition of the Mediterranean Diet; A Literature Review. Nutrients 2015, 7, 9139–9153.
- Khalifa, S.A.M.; Yosri, N.; El-Mallah, M.F.; Ghonaim, R.; Guo, Z.; Musharraf, S.G.; Du, M.; Khatib, A.; Xiao, J.; Saeed, A.; et al. Screening for natural and derived bio-active compounds in preclinical and clinical studies: One of the frontlines of fighting the coronaviruses pandemic. Phytomedicine 2021, 85, 153311.
- Yosri, N.; Abd El-Wahed, A.A.; Ghonaim, R.; Khattab, O.M.; Sabry, A.; Ibrahim, M.A.A.; Moustafa, M.F.; Guo, Z.; Zou, X.; Algethami, A.F.M.; et al. Anti-Viral and Immunomodulatory Properties of Propolis: Chemical Diversity, Pharmacological Properties, Preclinical and Clinical Applications, and In Silico Potential against SARS-CoV-2. Foods 2021, 10, 1776.
- World Health Organization. Weekly Epidemiological Update on COVID-19–22 February 2023, Edition 131, 22 February 2023. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid (accessed on 22 February 2023).
- da Silva, S.J.R.; do Nascimento, J.C.F.; Germano Mendes, R.P.; Guarines, K.M.; Targino Alves da Silva, C.; da Silva, P.G.; de Magalhães, J.J.F.; Vigar, J.R.J.; Silva-Júnior, A.; Kohl, A.; et al. Two Years into the COVID-19 Pandemic: Lessons Learned. ACS Infect. Dis. 2022, 8, 1758–1814.
- Ma, J.; Gu, Y.; Xu, P. A Roadmap to Engineering Antiviral Natural Products Synthesis in Microbes. Curr. Opin. Biotechnol. 2020, 66, 140–149.
- Musarra-Pizzo, M.; Pennisi, R.; Ben-Amor, I.; Mandalari, G.; Sciortino, M.T. Antiviral Activity Exerted by Natural Products against Human Viruses. Viruses 2021, 13, 828.
- Praditya, D.; Kirchhoff, L.; Bruning, J.; Rachmawati, H.; Steinmann, J.; Steinmann, E. Anti-infective properties of the golden spice curcumin. Front. Microbiol. 2019, 10, 912.
- Ikeda, K.; Tsujimoto, K.; Uozaki, M.; Nishide, M.; Suzuki, Y.; Koyama, A.H.; Yamasaki, H. Inhibition of multiplication of herpes simplex virus by caffeic acid. Int. J. Mol. Med. 2011, 28, 595–598.
- Treml, J.; Gazdová, M.; Šmejkal, K.; Šudomová, M.; Kubatka, P.; Hassan, S.T.S. Natural products-derived chemicals: Breaking barriers to novel anti-HSV drug development. Viruses 2020, 12, 154.
- Ojha, D.; Das, R.; Sobia, P.; Dwivedi, V.; Ghosh, S.; Samanta, A.; Chattopadhyay, D. Pedilanthus tithymaloides Inhibits HSV infection by modulating NF-κB signaling. PLoS ONE 2015, 10, e0139338.
- Wang, L.; Song, J.; Liu, A.; Xiao, B.; Li, S.; Wen, Z.; Lu, Y.; Du, G. Research Progress of the Antiviral Bioactivities of Natural Flavonoids. Nat. Prod. Bioprospect. 2020, 10, 271–283.
- Pandey, P.; Rane, J.S.; Chatterjee, A.; Kumar, A.; Khan, R.; Prakash, A.; Ray, S. Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: An in silico study for drug development. J. Biomol. Struct. Dyn. 2020, 22, 1–11.
- Yarmolinsky, L.; Huleihel, M.; Zaccai, M.; Ben-Shabat, S. Potent antiviral flavone glycosides from Ficus benjamina leaves. Fitoterapia 2012, 83, 362–367.
- Lee, S.; Lee, H.H.; Shin, Y.S.; Kang, H.; Cho, H. The anti-HSV-1 effect of quercetin is dependent on the suppression of TLR-3 in Raw 264.7 cells. Arch. Pharmacal. Res. 2017, 40, 623–630.
- Xu, J.; Xu, Z.; Zheng, W. A Review of the Antiviral Role of Green Tea Catechins. Molecules 2017, 22, 1337.
- Annunziata, G.; Maisto, M.; Schisano, C.; Ciampaglia, R.; Narciso, V.; Tenore, G.C.; Novellino, E. Resveratrol as a novel antiherpes simplex virus nutraceutical agent: An overview. Viruses 2018, 10, 473.
- Kesharwani, A.; Polachira, S.K.; Nair, R.; Agarwal, A.; Mishra, N.N.; Gupta, S.K. Anti-HSV-2 activity of Terminalia chebula Retz extract and its constituents, chebulagic and chebulinic acids. BMC Complem. Altern. Med. 2017, 17, 110.
- Rouf, R.; Uddin, S.J.; Sarker, D.K.; Islam, M.T.; Ali, E.S.; Shilpi, J.A.; Nahar, L.; Tiralongo, E.; Sarker, S.D. Antiviral potential of garlic (Allium sativum) and its organosulfur compounds: A systematic update of pre-clinical and clinical data. Trends Food Sci. Technol. 2020, 104, 219–234.
- Asif, M.; Saleem, M.; Saadullah, M.; Yaseen, H.S.; Al Zarzour, R. COVID-19 and therapy with essential oils having antiviral, anti-inflammatory, and immunomodulatory properties. Inflammopharmacology 2020, 28, 1153–1161.
- Shen, J.; Wang, G.; Zuo, J. Caffeic acid inhibits HCV replication via induction of IFNα antiviral response through p62-mediated Keap1/Nrf2 signaling pathway. Antivir. Res. 2018, 154, 166–173.
- Hsu, W.-C.; Chang, S.-P.; Lin, L.-C.; Li, C.-L.; Richardson, C.D.; Lin, C.-C.; Lin, L.-T. Limonium Sinense and gallic acid suppress hepatitis C virus infection by blocking early viral entry. Antivir. Res. 2015, 118, 139–147.
- Bose, M.; Kamra, M.; Mullick, R.; Bhattacharya, S.; Das, S.; Karande, A.A. Identification of a flavonoid isolated from plum (Prunus domestica) as a potent inhibitor of Hepatitis C virus entry. Sci. Rep. 2017, 7, 3965.
- Mishra, R.C.; Kumari, R.; Yadav, S.; Yadav, J.P. Antiviral potential of phytoligands against chymotrypsin-like protease of COVID-19 virus using molecular docking studies: An optimistic approach. Res. Square, 2020; ahead of print.
- Mahn, A.; Castillo, A. Potential of sulforaphane as a natural immune system enhancer: A review. Molecules 2021, 26, 752.
- Lynch, J.P., 3rd; Kajon, A.E. Adenovirus: Epidemiology, global spread of novel serotypes, and advances in treatment and prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602.
- Chiang, L.C.; Chiang, W.; Chang, M.Y.; Ng, L.T.; Lin, C.C. Antiviral activity of Plantago major extracts and related compounds in vitro. Antivir. Res. 2002, 55, 53–62.
- Utsunomiya, H.; Ichinose, M.; Ikeda, K.; Uozaki, M.; Morishita, J.; Kuwahara, T.; Koyama, A.H.; Yamasaki, H. Inhibition by caffeic acid of the influenza A virus multiplication in vitro. Int. J. Mol. Med. 2014, 34, 1020–1024.
- Li, Y.; Wang, L.; Si, H.; Yu, Z.; Tian, S.; Xiang, R.; Deng, X.; Liang, R.; Jiang, S.; Yu, F. Influenza virus glycoprotein-reactive human monoclonal antibodies. Microbes Infect. 2020, 22, 263–271.
- Lee, J.H.; Oh, M.; Seok, J.H.; Kim, S.; Lee, D.B.; Bae, G.; Bae, H.I.; Bae, S.Y.; Hong, Y.M.; Kwon, S.O.; et al. Antiviral Effects of Black Raspberry (Rubus coreanus) Seed and Its Gallic Acid against Influenza Virus Infection. Viruses 2016, 8, 157.
- Yamada, K.; Ogawa, H.; Hara, A.; Yoshida, Y.; Yonezawa, Y.; Karibe, K.; Nghia, V.B.; Yoshimura, H.; Yamamoto, Y.; Yamada, M.; et al. Mechanism of the antiviral effect of hydroxytyrosol on influenza virus appears to involve morphological change of the virus. Antivir. Res. 2009, 83, 35–44.
- Yan, H.; Wang, H.; Ma, L.; Ma, X.; Yin, J.; Wu, S.; Huang, H.; Li, Y. Cirsimaritin inhibits influenza A virus replication by downregulating the NF-κB signal transduction pathway. Virol. J. 2018, 15, 88.
- Lane, T.; Anantpadma, M.; Freundlich, J.S.; Davey, R.A.; Madrid, P.B.; Ekins, S. The Natural Product Eugenol Is an Inhibitor of the Ebola Virus In vitro. Pharm. Res. 2019, 36, 104.
- Dayem, A.A.; Choi, H.Y.; Kim, Y.B.; Cho, S.-G. Antiviral Effect of Methylated Flavonol Isorhamnetin against Influenza. PLoS ONE 2015, 10, e0121610.
- Sang, H.; Huang, Y.; Tian, Y.; Liu, M.; Chen, L.; Li, L.; Liu, S.; Yang, J. Multiple modes of action of myricetin in influenza A virus infection. Phytother. Res. 2021, 35, 2797–2806.
- Wu, W.; Li, R.; Li, X.; He, J.; Jiang, S.; Liu, S.; Yang, J. Quercetin as an antiviral agent inhibits influenza A virus (IAV) entry. Viruses 2015, 8, 6.
- Choi, H.J.; Song, J.H.; Park, K.S.; Kwon, D.H. Inhibitory effects of quercetin 3-rhamnoside on influenza A virus replication. Eur. J. Pharm. Sci. 2009, 37, 329–333.
- Sahoo, M.; Jena, L.; Rath, S.N.; Kumar, S. Identification of Suitable Natural Inhibitor against Influenza A (H1N1) Neuraminidase Protein by Molecular Docking. Genom. Inf. 2016, 14, 96–103.
- Bacelar Júnior, A.J.; Vieira Andrade, A.L.; Gomes Ferreira, A.A.; Andrade Oliveira, S.M.; Silva Pinheiro, T. Human Immunodeficiency Virus—HIV: A Review. Braz. J. Surg. Clin. Res. 2015, 9, 43–48.
- Nutan, M.M.; Goel, T.; Das, T.; Malik, S.; Suri, S.; Rawat, A.K.; Srivastava, S.K.; Tuli, R.; Malhotra, S.; Gupta, S.K. Ellagic acid & gallic acid from Lagerstroemia speciosa L. inhibit HIV-1 infection through inhibition of HIV-1 protease & reverse transcriptase activity. Ind. J. Med. Res. 2013, 137, 540–548.
- Bertelli, M.; Kiani, A.K.; Paolacci, S.; Manara, E.; Kurti, D.; Dhuli, K.; Bushati, V.; Miertus, J.; Pangallo, D.; Baglivo, M.; et al. Hydroxytyrosol: A natural compound with promising pharmacological activities. J. Biotechnol. 2020, 309, 29–33.
- Daino, G.L.; Frau, A.; Sanna, C.; Rigano, D.; Distinto, S.; Madau, V.; Esposito, F.; Fanunza, E.; Bianco, G.; Taglialatela-Scafati, O.; et al. Identification of Myricetin as an Ebola virus VP35-double-stranded RNA interaction inhibitor through a novel fluorescence-based assay. Biochemistry 2018, 57, 6367–6378.
- Ortega, J.T.; Suárez, A.I.; Serrano, M.L.; Baptista, J.; Pujol, F.H.; Rangel, H.R. The role of the glycosyl moiety of myricetin derivatives in anti-HIV-1 activity in vitro. AIDS Res. Ther. 2017, 14, 57.
- Kalita, R.; Bhattacharya, K.; Ali, A.; Sandilya, S. Quercitin as an antiviral weapon-A review. J. Appl. Pharm. Res. 2021, 9, 1–7.
- Choi, H.J.; Song, J.H.; Park, K.S.; Baek, S.H. In vitro anti-enterovirus 71 activity of gallic acid from Woodfordia fruticosa flowers. Lett. Appl. Microb. 2010, 50, 438–440.
- Zhang, L.; Li, Y.; Gu, Z.; Wang, Y.; Shi, M.; Ji, Y.; Sun, J.; Xu, X.; Zhang, L.; Jiang, J.; et al. Resveratrol Inhibits Enterovirus 71 Replication and Pro-Inflammatory Cytokine Secretion in Rhabdosarcoma Cells through Blocking IKKs/NFκB Signaling Pathway. PLoS ONE 2015, 10, e0116879.
- Yang, Y.; Xiu, J.; Liu, J.; Zhang, L.; Li, X.; Xu, Y.; Qin, C.; Zhang, L. Chebulagic Acid, a Hydrolyzable Tannin, Exhibited Antiviral Activity in vitro and in Vivo against Human Enterovirus 71. Int. J. Mol. Sci. 2013, 14, 9618–9627.
- Yang, Y.; Xiu, J.; Zhang, L.; Qin, C.; Liu, J. Antiviral activity of punicalagin toward human enterovirus 71 in vitro and in vivo. Phytomedicine 2012, 20, 67–70.
- Chapman, R.L.; Andurkar, S.V. A review of natural products, their effects on SARS-CoV-2 and their utility as lead compounds in the discovery of drugs for the treatment of COVID-19. Med. Chem. Res. 2022, 31, 40–51.
- Keflie, T.S.; Biesalski, H.K. Micronutrients and bioactive substances: Their potential roles in combating COVID-19. Nutrition 2021, 84, 111103.
- Alrasheid, A.A.; Babiker, M.Y.; Awad, T.A. Evaluation of certain medicinal plants compounds as new potential inhibitors of novel corona virus (COVID-19) using molecular docking analysis. In silico. Pharmacology 2021, 9, 10.
- Shaldam, M.A.; Yahya, G.; Mohamed, N.H.; Abdel-Daim, M.M.; Al Naggar, Y. In silico Screening of Potent Bioactive Compounds from Honey Bee Products Against COVID-19 Target Enzymes. Environ. Sci. Pollut. Res. 2021, 28, 40507–40514.
- Omotuyi, I.O.; Nash, O.; Ajiboye, B.O.; Olumekun, V.O.; Oyinloye, B.E.; Olonisakin, A.; Ajayi, A.O.; Olusanya, O.; Akomolafe, F.S.; Adelakun, N. Aframomum melegueta secondary metabolites exhibit polypharmacology against SARS-CoV-2 drug targets: In vitro validation of furin inhibition. Phyther. Res. 2020, 35, 908–919.
- Khaerunnisa, S.; Kurniawan, H.; Awaluddin, R.; Suhartati, S.; Soetjipto, S. Potential Inhibitor of COVID-19 Main Protease (Mpro) From Several Medicinal Plant Compounds by Molecular Docking Study. Preprints 2020, 2020030226.
- Bilginer, S.; Gözcü, S.; Güvenalp, Z. Molecular Docking Study of Several Seconder Metabolites from Medicinal Plants as Potential Inhibitors of COVID-19 Main Protease. Turk. J. Pharm. Sci. 2022, 19, 431–441.
- Halder, P.; Pal, U.; Paladhi, P.; Dutta, S.; Paul, P.; Pal, S.; Das, D.; Ganguly, A.; Dutta, I.; Mandal, S.; et al. Evaluation of potency of the selected bioactive molecules from Indian medicinal plants with MPro of SARS-CoV-2 through in silico analysis. J. Ayurveda Integr. Med. 2022, 13, 100449.
- Mukherjee, S.; Sharma, D.; Sharma, A.K.; Jaiswal, S.; Sharma, N.; Borah, S.; Kaur, G. Flavan-based phytoconstituents inhibit Mpro, a SARS-COV-2 molecular target, in silico. J. Biomol. Struct. Dyn. 2022, 40, 11545–11559.
- Rakshit, G.; Dagur, P.; Satpathy, S.; Patra, A.; Jain, A.; Ghosh, M. Flavonoids as potential therapeutics against novel coronavirus disease-2019 (nCOVID-19). J. Biomol. Struct. Dyn. 2022, 40, 6989–7001.
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.S. Inhibition of SARSCoV 3CL protease by flavonoids. J. Enzyme. Inhib. Med. Chem. 2020, 35, 145–151.
- Jo, S.; Kim, S.; Kim, D.Y.; Kim, M.S.; Shin, D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzyme Inhib. Med. Chem. 2020, 35, 1539–1544.
- Vicidomini, C.; Roviello, V.; Roviello, G.N. In silico Investigation on the Interaction of Chiral Phytochemicals from Opuntia ficus-indica with SARS-CoV-2 Mpro. Symmetry 2021, 13, 1041.
- Joshi, R.S.; Jagdale, S.S.; Bansode, S.B.; Shiva Shankar, S.; Tellis, M.B.; Pandya, V.K.; Chugh, A.; Giri, A.P.; Kulkarni, M.J. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J. Biomol. Struct. Dyn. 2020, 39, 3099–3114.
- Cherrak, S.A.; Merzouk, H.; Mokhtari-Soulimane, N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking and simulation studies. PLoS ONE 2020, 15, e0240653.
- Frengki, F.; Putra, D.P.; Wahyuni, F.S.; Khambri, D.; Vanda, H.; Sofia, V. Potential antiviral of catechins and their derivatives to inhibit sars-cov-2 receptors of Mpro protein and spike glycoprotein in COVID-19 through the in silico approach. J. Kedokt. Hewan 2020, 14, 59–65.
- Pandey, A.K.; Verma, S. An in-silico evaluation of dietary components for structural inhibition of SARS-Cov-2 main protease. J. Biomol. Struct. Dyn. 2022, 40, 136–142.
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Evaluation of green tea polyphenols as novel corona virus (SARS-CoV-2) main protease (Mpro) inhibitors—An in silico docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2021, 39, 4362–4374.
- Tallei, T.E.; Tumilaar, S.G.; Niode, N.J.; Fatimawali, F.; Kepel, B.J.; Idroes, R.; Effendi, Y.; Sakib, S.A.; Emran, T.B. Potential of Plant Bioactive Compounds as SARS-CoV-2 Main Protease (Mpro) and Spike (S) Glycoprotein Inhibitors: A Molecular Docking Study. Scientifica 2020, 2020, 6307457.
- Mhatre, S.; Naik, S.; Patravale, V. A molecular docking study of EGCG and theaflavin digallate with the druggable targets of SARS-CoV-2. Comput. Biol. Med. 2021, 129, 104137.
- Mostafa, N.M.; Ismail, M.I.; El-Araby, A.M.; Bahgat, D.M.; Elissawy, A.M.; Mostafa, A.M.; Eldahshan, O.A.; Singab, A.N.B. Investigation of SARS-CoV-2 Main Protease Potential Inhibitory Activities of Some Natural Antiviral Compounds Via Molecular Docking and Dynamics Approaches. Phyton Int. J. Exp. Bot. 2022, 91, 1089–1104.
- Rathinavel, T.; Palanisamy, M.; Palanisamy, S.; Subramanian, A.; Thangaswamy, S. Phytochemical 6-Gingerol—A promising Drug of choice for COVID-19. Int. J. Adv. Sci. Eng. 2020, 6, 1482–1489.
- Du, R.; Cooper, L.; Chen, Z.; Lee, H.; Rong, L.; Cui, Q. Discovery of chebulagic acid and punicalagin as novel allosteric inhibitors of SARS-CoV-2 3CLpro. Antivir. Res. 2021, 190, 105075.
- Hussain, T.; Habib, A.H.; Rafeeq, M.M.; Alafnan, A.; Khafagy, E.-S.; Iqbal, D.; Jamal, Q.M.S.; Unissa, R.; Sharma, D.C.; Moin, A.; et al. Oleuropein as a Potent Compound against Neurological Complications Linked with COVID-19: A Computational Biology Approach. Entropy 2022, 24, 881.
- Abdizadeh, R.; Hadizadeh, F.; Abdizadeh, T. In silico analysis and identification of antiviral coumarin derivatives against 3-chymotrypsin-like main protease of the novel coronavirus SARS-CoV-2. Mol. Divers 2022, 26, 1053–1076.
- Shekh, S.; Reddy, K.K.A.; Gowd, K.H. In silico allicin induced S-thioallylation of SARS-CoV-2 main protease. J. Sulf. Chem. 2021, 42, 109–120.
- Paolacci, S.; Kiani, A.K.; Shree, P.; Tripathi, D.; Tripathi, Y.B.; Tripathi, P.; Tartaglia, G.M.; Farronato, M.; Farronato, G.; Connelly, S.T.; et al. Scoping review on the role and interactions of hydroxytyrosol and alpha-cyclodextrin in lipid-raft-mediated endocytosis of SARS-CoV-2 and bioinformatic molecular docking studies. Eur. Rev. Med. Pharmacol. Sci. 2021, 25 (Suppl. 1), 90–100.
- Sekiou, O.; Bouziane, I.; Frissou, N.; Bouslama, Z.; Honcharova, O.; Djemel, A.; Benselhoub, A. In-Silico Identification of Potent Inhibitors of COVID-19 Main Protease (Mpro) from Natural Products. Int. J. Biochem. Physiol. 2020, 5, 16000189.
- Bhowmik, D.; Nandi, R.; Jagadeesan, R.; Kumar, N.; Prakash, A.; Kumar, D. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infect. Genet. Evol. 2020, 84, 104451.
- Orfali, R.; Rateb, M.E.; Hassan, H.M.; Alonazi, M.; Gomaa, M.R.; Mahrous, N.; GabAllah, M.; Kandeil, A.; Perveen, S.; Abdelmohsen, U.R.; et al. Sinapic Acid Suppresses SARS-CoV-2 Replication by Targeting Its Envelope Protein. Antibiotics 2021, 10, 420.
- Muhammad, S.; Hira, S.; Al-sehemi, A.G.; Abdullah, H.; Khan, M.; Irfan, M.; Iqbal, J. Exploring the new potential antiviral constituents of Moringa oliefera for SARS-CoV-2 pathogenesis: An in silico molecular docking and dynamic studies. Chem. Phys. Lett. 2021, 767, 138379.
- Pitsillou, E.; Liang, J.; Ververis, K.; Lim, K.W.; Hung, A.; Karagiannis, T.C. Identification of Small Molecule Inhibitors of the Deubiquitinating Activity of the SARS-CoV-2 Papain-Like Protease: In silico Molecular Docking Studies and in vitro Enzymatic Activity Assay. Front. Chem. 2020, 8, 623971.
- Suručić, R.; Tubić, B.; Stojiljković, M.P.; Djuric, D.M.; Travar, M.; Grabež, M.M.; Šavikin, K.; Škrbić, R. Computational study of pomegranate peel extract polyphenols as potential inhibitors of SARS-CoV-2 virus internalization. Mol. Cell. Biochem. 2021, 476, 1179–1193.
- Özdemir, M.; Köksoy, B.; Ceyhan, D.; Sayın, K.; Erçağ, E.; Bulut, M.; Yalçın, B. Design and in silico study of the novel coumarin derivatives against SARS-CoV-2 main enzymes. J. Biomol. Struct. Dyn. 2020, 27, 1–16.
This entry is offline, you can click here to edit this entry!