Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Since the discovery of insulin a century ago, insulin injection has been a primary treatment for both type 1 (T1D) and type 2 diabetes (T2D). T2D is a complicated disease that is triggered by the dysfunction of insulin-producing β cells and insulin resistance in peripheral tissues. Insulin injection partially compensates for the role of endogenous insulin which promotes glucose uptake, lipid synthesis and organ growth.
- diabetes
- stem cells
- human islet-like organoids
1. Introduction
Diabetes encompasses metabolic disorders that lead to hyperglycemia and is caused by various factors such as infection, autoimmunity, obesity, and stresses [1][2][3]. Diabetes-induced hyperglycemia is linked with long-lasting co-morbidities such as hypertension, hyperlipidemia, chronic kidney disease, cardiovascular disease, retinopathy, and neuropathy [4][5]. As such, diabetes poses a significant global health risk with over 537 million individuals currently suffering from the disease and this led to 6.7 million deaths in 2021. According to the International Federation of Diabetes Atlas, the incidence of diabetes is projected to increase to over 783 million diabetic individuals by 2045 [3]. Diabetes is generally classified as either type 1 diabetes (T1D) or type 2 diabetes (T2D). Although both conditions result in a hyperglycemic condition, their etiology is markedly different.
T1D is characterized by an immune assault on the islet β cells [6]. The mechanism for T1D pathogenesis is highly heterogeneous but not fully understood yet. Multiple genetic and environmental factors can act as potential causes for immune autoreactivity against islet β cells. For instance, HLA-haplotype polymorphisms are involved in T1D susceptibility by influencing β cell interaction with immune cell populations. Variation in the INS gene can result in reduced insulin production in the thymus, thus preventing the detection and elimination of auto-reactive T-cells against β cells [7]. CFTR variants that hamper pancreatic exocrine function can result in increased inflammation, and thus greater T1D risk [8]. Additionally, changes in the microbiota, environmental conditions, or exposure to viruses or germs that exhibit molecular mimicry with endogenous factors of the body can trigger the production of autoantibodies against insulin and β cells [7].
Pancreatic islets isolated from T1D patients exhibit increased markers of ER stress as well as senescence and inflammatory signals, which lead to reduced expression of β cell identity markers such as insulin. The ER stress also disrupts the insulin:proinsulin ratio, which decreases the efficacy of glycemic control by insulin. This dysfunction is linked with the reduced β cell population and increased α cell population in T1D islets, leading to the dysregulation of glucagon secretion and accelerated blood sugar elevation [9][10].
T2D is by far the most common form of diabetes [11]. Modern diet and lifestyle habits have played an important role in exacerbating the prevalence of T2D [12][13]. T2D is becoming exceedingly burdening on the global healthcare systems with over 966 billion dollars spent [3]. The mechanism of T2D is not primarily involved with immune autoreactivity, but rather, β cell dysfunction and oxidative stress-induced inflammatory death in response to increased insulin resistance.
Diabetes treatment typically relies on insulin or insulin analog injections for maintaining normoglycemia. Although islet β cell dysfunction is considered the primary cause of hyperglycemia in both T1D and T2D, the recent advances in hPSC-derived islet systems, are currently being investigated mainly for T1D. hPSCs such as embryonic pluripotent stem cells (hESCs) [14] or human induced pluripotent stem cells (hiPSCs) [15] provide the opportunity to recapture human islet organogenesis and neogenesis in a scalable manner.
2. T2D Pathogenesis: From Insulin Resistance to β Cell Death
T2D is a progressive disease that can be broken down into four stages: (1) defect in β cell glucose-stimulated insulin secretion (GSIS); (2) peripheral insulin resistance; (3) β cell compensation; (4) β cell loss. Researchers discuss the progression of T2D and the potential advances in hPSC-derived islets (Figure 1).
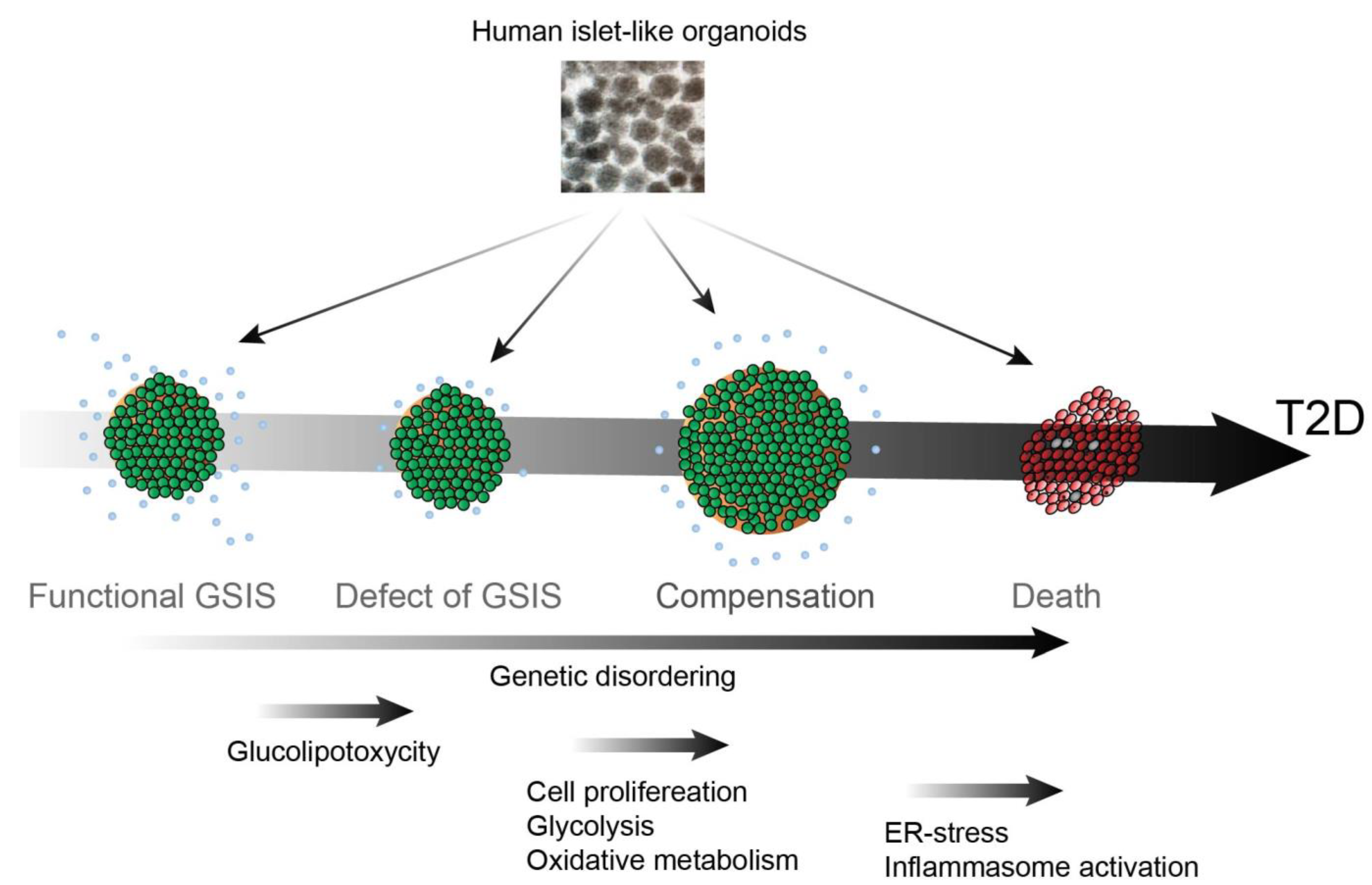
Figure 1. hPSC-derived islet for studying the pathogenesis of T2D.
2.1. Defect of GSIS
In healthy individuals, insulin secretion from functional β cells is an acute phase response. In the first phase, functional β cells secrete insulin within a few minutes of glucose stimulation followed by the second phase insulin secretion, in which a slower response is induced by continuous glucose stimulation. It was found that acute insulin secretory responses (AIR) to intravenous glucose are lower in individuals with impaired glucose tolerance and those at high risk of developing T2D [16]. Defects in the first phase of GSIS lead to inefficient glucose uptake in peripheral tissues and suppression of gluconeogenesis in the liver. Currently, glucagon-like peptide-1 (GLP-1) is one of the most popular targets for T2D treatment, as it is considered to enhance GSIS function in patients with glucose intolerance or T2D [17].
2.2. Insulin Resistance
T2D onset and progression is promoted by insulin resistance in peripheral tissues, particularly in the adipose, skeletal muscle, and hepatocyte tissues, and is often accompanied by obesity and related lipid dysregulation. Obesity can trigger chronic inflammation by recruiting macrophages in adipose tissues. Inflammation caused by macrophages, characterized by enhanced expression of cytokine markers such as nuclear factor kappa B (NF-κB) and tumor necrosis factor-alpha (TNF-α), induces insulin receptor resistance in adipocytes. Insulin is a strong suppressor of adipose lipolysis, therefore, defects of the insulin signaling in insulin resistance enhance lipolysis [18]. Partial inhibition of lipolysis through the inhibition of hormone-sensitive lipase has been found to result in a decrease in fatty acid influx into white adipose tissues, and an increase in glucose metabolism, lipogenesis, and insulin-mediated glucose uptake [19]. Insulin and fibroblast growth factor-1 (FGF-1) suppress lipolysis through phosphodiesterase 3B (PDE3B) and PDE4D, respectively [20]. Through inter-organ cross-talk, increased lipolysis at adipose tissues plays an important role in not only adipose insulin resistance but also the liver and skeletal muscle. Adipose lipolysis generates excess free fatty acids (FFAs). In 1985, Marchand-Brustel et al. showed that phosphorylation activity of the insulin receptor substrate 1 (IRS-1) in skeletal muscles was markedly reduced in obesity-induced diabetic mice [21]. Later, Griffin et al. showed that an increase in plasma-free fatty acids as a result of the infusion of lipids to mice undergoing a hyperinsulinemic-euglycemic clamp resulted in a significantly decreased glucose infusion rate [22]. The data also showed a significant decrease in skeletal insulin receptor signaling through attenuated tyrosine phosphorylation at insulin receptor substrate-1 (IRS-1) and AKT, which regulates Glut4 cycling and glucose uptake; further, a four-fold increase in protein kinase theta (PKC-θ) activity was observed. A follow-up study demonstrated that PKC-θ knockout mice did not exhibit such a decrease in insulin receptor activity and glucose uptake in response to fat accumulation within skeletal muscles [23]. Increased cytosolic diacylglycerols were observed upon post-lipid infusion induced PKC-θ mediated phosphorylation of serine residue 1101 on IRS-1, which inhibits the receptors’ tyrosine kinases [24]. Further studies have shown that lipids can also alter mitochondrial dynamics resulting in insulin insensitivity caused by lipid-induced mitochondrial fission [25]. A similar mechanism of insulin resistance has also been demonstrated in the liver, wherein increased diglycerides (DAGs) in response to lipid accumulation activate a different isoform of protein kinase C epsilon (PKCε), which in turn phosphorylates serine residues on IRS-2 and reduces glucose production and release in response to insulin signaling [26]. Thus, multi-organ insulin resistance increases the demand for insulin from β cells.
2.3. β Cell Compensation
Although defective GSIS and insulin insensitivity mark the initial stages of progression into T2D, patients can still maintain normoglycemia as long as the β cells exhibit adaptive functionality [27]. In response to insulin demand, β cells undergo proliferation as a compensatory defense mechanism against insulin resistance [28][29]. β cell function is known to exhibit a hyperbolic compensatory relationship with respect to insulin sensitivity, meaning that a decrease in insulin responsiveness within peripheral tissues is accompanied by a greater β cell insulin response. Patients that maintain this hyperbolic relationship can remain within normal glycemic levels. Patients whose β cells fail to maintain this compensatory relationship have been shown to develop glucose intolerance and ultimately T2D hyperglycemia [30]. Several studies have shown that a high-fat diet (HFD) induces glucose intolerance accompanied by insulin resistance, while still maintaining normoglycemia with an increased β cell mass and total insulin levels [31][32]. This β cell compensatory machinery in response to insulin resistance has also been observed in many prediabetic animal models [33][34]. However, the chronic hyperinsulinemia that results from the β cell compensation can, result in accelerated insulin resistance in peripheral tissues and eventually lead to β cell exhaustion and apoptosis.
2.4. β Cell Death
If the β cell compensatory mechanism fails, patients will no longer be able to maintain homeostatic glucose levels. Elevated postprandial glucose levels and glucose intolerance will eventually progress into full-blown diabetic hyperglycemia accompanied by β cell loss. Glucolipotoxicity, resulting from increased levels of plasma lipids and glucose has many other negative effects on β cells. Researchers have previously identified that thioredoxin interacting protein (TXNIP), originally called Vitamin D3 upregulated protein (VDUP1) or thioredoxin binding protein-2 (TBP-2) plays a central role in β cell dysfunction and death [35][36][37][38][39]. TXNIP has a reciprocal function with the antioxidant protein Thioredoxin (TRX); thus, increased TXNIP expression is linked with enhanced oxidative stress [37][38][39]. TXNIP interacts with NLRP3, a component of the inflammasome to activate IL1β expression in islets, thus suggesting the mechanism of cytokine-induced β cell dysfunction in T2D [40]. Another leading hypothesis for β cell dysfunction is that the increased need for insulin production to counteract peripheral insulin resistances places exorbitant endoplasmic reticulum (ER) stress on β cells. Increased ER stress then activates the unfolded protein response (UPR) pathway, which in turn, can activate NF-κB to trigger β cell apoptosis [9][41][42]. ER stress was found to induce TXNIP expression; thus, TXNIP also plays a central role in ER stress-mediated inflammasome activation and β cell apoptosis in T2D [43]. ER stress also contributes to the initiation of islet amyloid polypeptide (IAPP) accumulation by stimulating protein misfolding in β cells, which further induces Chop-mediated cell death [44]. Masters et al. found that increased IAPP production in β cells activates the NLRP3 inflammasome, leading to inflammation and macrophage recruitment. Human IAPP (hIAPP) but not rat or mouse IAPP shows toxicity in β cells by an accumulation of hIAPP dimers and more complex amyloid oligomer formation, triggering an autophagic response to restrict β cell mass [45][46]. T2D is often accompanied by aging, with an enhanced glycolytic and reduced oxidative metabolic signature, which contributes to reducing glucose sensitivity in aged diabetic β cells, as insulin secretion is tightly regulated by metabolic function [47][48]. Aguayo-Mazzucato et al. further found that aging in conjunction with insulin resistance both contribute to β cells that exhibit increased markers of senescence. These senescent β cells are resistant to apoptosis and secrete senescence-associated secretory phenotype (SASP) factors such as inflammatory cytokines, proteases, and DNA fragments, which further induce dysfunction in the surrounding β cells. Increased senolysis through oral ABT263 has been found to improve β cell metabolic function and reduce T2D symptoms. Other metabolic inquiries such as those by Lupse et al. have found that PHLP phosphatase inhibition can protect β cells from diabetic death [49]. Hyperglycemic stress chronically activates the mTORC1 pathway, which in turn upregulates protein phosphatase PHLPP-like protein (PHLP) pathways. PHLP phosphatases will deactivate the cellular signals for β cell survival such as AKT which in turn will trigger β cell apoptosis and contribute to T2D development.
In addition to changes in metabolic function, apoptotic rate, insulin production and processing and inflammation, β cells have been shown to undergo dedifferentiation in response to hyperglycemic stress. Lineage tracing experiments in mice have revealed that β cells dedifferentiate into a progenitor-like state as exemplified by the upregulation of pluripotency markers, such as NANOG and OCT4 [50]. Furthermore, dedifferentiation often leads to the neogenesis of other hormonal cells including glucagon-producing α cells in T2D patients [51]. Sox5 knockdown significantly reduces GSIS and calcium-induced exocytotic depolarization, whereas Sox5 overexpression enhances the gene expression necessary for β cell identity, thus improving insulin secretion by β cells in mouse T2D models. Oxidative stress has been suggested as a leading factor for ectopic β cell fates, with increased oxidative mitochondrial stress being associated with upregulated JNK pathways and downregulated β cell identity genes such as FOXO1, MAFA, and PDX1 [52][53][54][55][56]. Oxidative stress in response to glucolipotoxicity has also been shown to result in increased ROS production through NADPH oxidase (NOX), which in turn impacts β cell failure through aberrant changes in the mTOR pathways that affect β cell insulin production, β cell mass, proliferation, dedifferentiation and mitochondria-mediated apoptosis [57][58].
T2D progression is accelerated by insulin resistance but is critically dependent on β cell failure. Therefore, it is important to understand the stage-specific event of β cells during T2D pathogenesis. Prior to β cell death, diet as well as exercise, supplemented with insulin sensitizers may reverse a prediabetic state with β cell restoration. However, once full T2D has manifested through β cell death, the most important long-term solution is β cell regeneration or replacement. This is supported by recent studies, which demonstrated that a decrease in visceral fat is only able to induce diabetic remission if β cells maintain a capacity for functional recovery [59]. Following these aspects, researchers suggest the hPSC-derived islets can be used for screening the drugs effectively restores β cells in the early stage of T2D, whereas transplantation of hPSC-derived islets is a new therapeutic for late-stage of T2D.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23095099
References
- Huang, Y.; Karuranga, S.; Malanda, B.; Williams, D.R.R. Call for data contribution to the IDF Diabetes Atlas 9th Edition 2019. Diabetes Res. Clin. Pract. 2018, 140, 351–352.
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281.
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119.
- Schuster, D.P.; Duvuuri, V. Diabetes mellitus. Clin. Podiatr. Med. Surg. 2002, 19, 79–107.
- Kimura, A.; Toyoda, T.; Nishi, Y.; Nasu, M.; Ohta, A.; Osafune, K. Small molecule AT7867 proliferates PDX1-expressing pancreatic progenitor cells derived from human pluripotent stem cells. Stem Cell Res. 2017, 24, 61–68.
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the beta-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 2021, 17, 150–161.
- Ilonen, J.; Lempainen, J.; Veijola, R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 635–650.
- Chiou, J.; Geusz, R.J.; Okino, M.L.; Han, J.Y.; Miller, M.; Melton, R.; Beebe, E.; Benaglio, P.; Huang, S.; Korgaonkar, K.; et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 2021, 594, 398–402.
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008, 29, 42–61.
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362.
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111.
- Sami, W.; Ansari, T.; Butt, N.S.; Hamid, M.R.A. Effect of diet on type 2 diabetes mellitus: A review. Int. J. Health Sci. 2017, 11, 65–71.
- Jannasch, F.; Kroger, J.; Schulze, M.B. Dietary Patterns and Type 2 Diabetes: A Systematic Literature Review and Meta-Analysis of Prospective Studies. J. Nutr. 2017, 147, 1174–1182.
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147.
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872.
- Pratley, R.E.; Weyer, C. The role of impaired early insulin secretion in the pathogenesis of Type II diabetes mellitus. Diabetologia 2001, 44, 929–945.
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157.
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22.
- Girousse, A.; Tavernier, G.; Valle, C.; Moro, C.; Mejhert, N.; Dinel, A.L.; Houssier, M.; Roussel, B.; Besse-Patin, A.; Combes, M.; et al. Partial inhibition of adipose tissue lipolysis improves glucose metabolism and insulin sensitivity without alteration of fat mass. PLoS Biol. 2013, 11, e1001485.
- Sancar, G.; Liu, S.; Gasser, E.; Alvarez, J.G.; Moutos, C.; Kim, K.; van Zutphen, T.; Wang, Y.; Huddy, T.F.; Ross, B.; et al. FGF1 and insulin control lipolysis by convergent pathways. Cell Metab. 2022, 34, 171–183.e6.
- Le Marchand-Brustel, Y.; Gremeaux, T.; Ballotti, R.; Van Obberghen, E. Insulin receptor tyrosine kinase is defective in skeletal muscle of insulin-resistant obese mice. Nature 1985, 315, 676–679.
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274.
- Kim, J.K.; Fillmore, J.J.; Sunshine, M.J.; Albrecht, B.; Higashimori, T.; Kim, D.W.; Liu, Z.X.; Soos, T.J.; Cline, G.W.; O’Brien, W.R.; et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J. Clin. Investig. 2004, 114, 823–827.
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Muller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCtheta in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602.
- Axelrod, C.L.; Fealy, C.E.; Erickson, M.L.; Davuluri, G.; Fujioka, H.; Dantas, W.S.; Huang, E.; Pergola, K.; Mey, J.T.; King, W.T.; et al. Lipids activate skeletal muscle mitochondrial fission and quality control networks to induce insulin resistance in humans. Metabolism 2021, 121, 154803.
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91.
- Kahn, S.E.; Prigeon, R.L.; McCulloch, D.K.; Boyko, E.J.; Bergman, R.N.; Schwartz, M.W.; Neifing, J.L.; Ward, W.K.; Beard, J.C.; Palmer, J.P.; et al. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 1993, 42, 1663–1672.
- Linnemann, A.K.; Baan, M.; Davis, D.B. Pancreatic beta-cell proliferation in obesity. Adv. Nutr. 2014, 5, 278–288.
- Sharma, R.B.; O’Donnell, A.C.; Stamateris, R.E.; Ha, B.; McCloskey, K.M.; Reynolds, P.R.; Arvan, P.; Alonso, L.C. Insulin demand regulates beta cell number via the unfolded protein response. J. Clin. Investig. 2015, 125, 3831–3846.
- Kahn, S.E. Clinical review 135: The importance of beta-cell failure in the development and progression of type 2 diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 4047–4058.
- Gonzalez, A.; Merino, B.; Marroqui, L.; Neco, P.; Alonso-Magdalena, P.; Caballero-Garrido, E.; Vieira, E.; Soriano, S.; Gomis, R.; Nadal, A.; et al. Insulin hypersecretion in islets from diet-induced hyperinsulinemic obese female mice is associated with several functional adaptations in individual beta-cells. Endocrinology 2013, 154, 3515–3524.
- Irles, E.; Neco, P.; Lluesma, M.; Villar-Pazos, S.; Santos-Silva, J.C.; Vettorazzi, J.F.; Alonso-Magdalena, P.; Carneiro, E.M.; Boschero, A.C.; Nadal, A.; et al. Enhanced glucose-induced intracellular signaling promotes insulin hypersecretion: Pancreatic beta-cell functional adaptations in a model of genetic obesity and prediabetes. Mol. Cell Endocrinol. 2015, 404, 46–55.
- Cavaghan, M.K.; Ehrmann, D.A.; Polonsky, K.S. Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J. Clin. Investig. 2000, 106, 329–333.
- Kang, T.; Boland, B.B.; Jensen, P.; Alarcon, C.; Nawrocki, A.; Grimsby, J.S.; Rhodes, C.J.; Larsen, M.R. Characterization of Signaling Pathways Associated with Pancreatic beta-cell Adaptive Flexibility in Compensation of Obesity-linked Diabetes in db/db Mice. Mol. Cell Proteom. 2020, 19, 971–993.
- Oka, S.; Yoshihara, E.; Bizen-Abe, A.; Liu, W.; Watanabe, M.; Yodoi, J.; Masutani, H. Thioredoxin binding protein-2/thioredoxin-interacting protein is a critical regulator of insulin secretion and peroxisome proliferator-activated receptor function. Endocrinology 2009, 150, 1225–1234.
- Yoshihara, E.; Fujimoto, S.; Inagaki, N.; Okawa, K.; Masaki, S.; Yodoi, J.; Masutani, H. Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nat. Commun. 2010, 1, 127.
- Yoshihara, E.; Chen, Z.; Matsuo, Y.; Masutani, H.; Yodoi, J. Thiol redox transitions by thioredoxin and thioredoxin-binding protein-2 in cell signaling. Methods Enzym. 2010, 474, 67–82.
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014, 4, 514.
- Yoshihara, E. TXNIP/TBP-2: A Master Regulator for Glucose Homeostasis. Antioxidants 2020, 9, 765.
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140.
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110.
- Donath, M.Y.; Ehses, J.A.; Maedler, K.; Schumann, D.M.; Ellingsgaard, H.; Eppler, E.; Reinecke, M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 2005, 54 (Suppl. 2), S108–S113.
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273.
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027.
- Kim, J.; Park, K.; Kim, M.J.; Lim, H.; Kim, K.H.; Kim, S.W.; Lee, E.S.; Kim, H.H.; Kim, S.J.; Hur, K.Y.; et al. An autophagy enhancer ameliorates diabetes of human IAPP-transgenic mice through clearance of amyloidogenic oligomer. Nat. Commun. 2021, 12, 183.
- Kim, J.; Cheon, H.; Jeong, Y.T.; Quan, W.; Kim, K.H.; Cho, J.M.; Lim, Y.M.; Oh, S.H.; Jin, S.M.; Kim, J.H.; et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. J. Clin. Investig. 2014, 124, 3311–3324.
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic beta-cells. Nat. Commun. 2019, 10, 2474.
- Murao, N.; Yokoi, N.; Takahashi, H.; Hayami, T.; Minami, Y.; Seino, S. Increased glycolysis affects beta-cell function and identity in aging and diabetes. Mol. Metab. 2022, 55, 101414.
- Lupse, B.; Annamalai, K.; Ibrahim, H.; Kaur, S.; Geravandi, S.; Sarma, B.; Pal, A.; Awal, S.; Joshi, A.; Rafizadeh, S.; et al. Inhibition of PHLPP1/2 phosphatases rescues pancreatic beta-cells in diabetes. Cell Rep. 2021, 36, 109490.
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234.
- Axelsson, A.S.; Mahdi, T.; Nenonen, H.A.; Singh, T.; Hanzelmann, S.; Wendt, A.; Bagge, A.; Reinbothe, T.M.; Millstein, J.; Yang, X.; et al. Sox5 regulates beta-cell phenotype and is reduced in type 2 diabetes. Nat. Commun. 2017, 8, 15652.
- Leenders, F.; Groen, N.; de Graaf, N.; Engelse, M.A.; Rabelink, T.J.; de Koning, E.J.P.; Carlotti, F. Oxidative Stress Leads to beta-Cell Dysfunction Through Loss of beta-Cell Identity. Front. Immunol. 2021, 12, 690379.
- Maechler, P.; Jornot, L.; Wollheim, C.B. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J. Biol. Chem. 1999, 274, 27905–27913.
- Kaneto, H.; Xu, G.; Fujii, N.; Kim, S.; Bonner-Weir, S.; Weir, G.C. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem. 2002, 277, 30010–30018.
- Harmon, J.S.; Stein, R.; Robertson, R.P. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J. Biol. Chem. 2005, 280, 11107–11113.
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid Redox Signal 2017, 26, 501–518.
- Vilas-Boas, E.A.; Almeida, D.C.; Roma, L.P.; Ortis, F.; Carpinelli, A.R. Lipotoxicity and beta-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress. Cells 2021, 10, 3328.
- Eguchi, N.; Vaziri, N.D.; Dafoe, D.C.; Ichii, H. The Role of Oxidative Stress in Pancreatic beta Cell Dysfunction in Diabetes. Int. J. Mol. Sci. 2021, 22, 1509.
- Taylor, R.; Al-Mrabeh, A.; Zhyzhneuskaya, S.; Peters, C.; Barnes, A.C.; Aribisala, B.S.; Hollingsworth, K.G.; Mathers, J.C.; Sattar, N.; Lean, M.E.J. Remission of Human Type 2 Diabetes Requires Decrease in Liver and Pancreas Fat Content but Is Dependent upon Capacity for beta Cell Recovery. Cell Metab. 2018, 28, 547–556.e3.
This entry is offline, you can click here to edit this entry!