2. HDL/apoA-I Kinetics
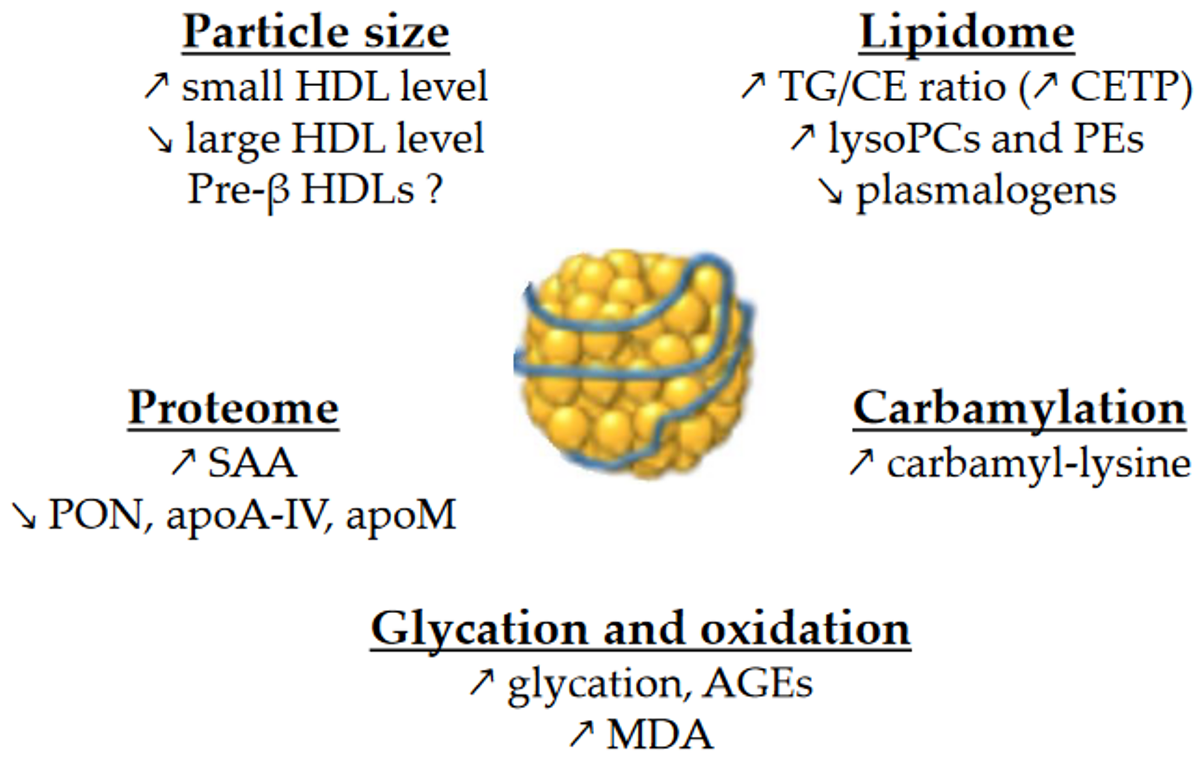
Numerous in vivo kinetic studies demonstrate that low HDL-C levels in T2DM and obesity are due to an accelerated catabolism of HDL particles [13][14][33][34][35]. The fractional catabolic rate (FCR) of HDL-apoA-I is increased, resulting in shorter plasma residence times for HDL particles [9][33][36]. Obese patients at an early stage of insulin resistance (i.e., without impaired glucose tolerance) already have an accelerated HDL-apoA-I catabolism [37].
Studies have found the production rate (PR) of HDL-apoA-I to be either elevated [9][13][34][36] or normal [14][37] in insulin-resistant individuals. The PR of HDL-cholesterol has recently been reported to be higher in patients with T2DM. In any case, the increase in HDL-apoA-I PR is usually smaller than the increase in the FCR, which explains the decreased concentrations of HDL-apoA-I. Interestingly, chronic endogenous hyperinsulinemia without insulin resistance (patients with insulinoma) does not induce an increase in the HDL-apoA-I PR [13]. This suggests that hyperinsulinemia per se is not responsible for the increased PR of HDL-apoA-I that is sometimes reported in patients with insulin resistance.
Glycation and glycoxidation may play a role in the accelerated catabolism of HDLs, although there is reportedly no correlation between HDL-apoA-I FCR and HbA1c [35]. Thus, the turnover of glycated apoA-I is almost three times faster than its non-glycated counterpart [36]. In addition, the methylglyoxal modification of HDLs reduces plasma half-life in vivo [23]. Yet considerable evidence suggests that the kinetic properties of HDLs are linked to TG metabolism. For instance, the HDL-apoA-I FCR in obese individuals is associated with triglyceridemia [38][39], VLDL-apoB PR [38], VLDL1-TG FCR and PR [39], and also with the HDL-TG/CE ratio [37]. In T2DM, the HDL-apoA-I FCR is also positively associated with plasma TG [35] and with the TG content in HDLs [14][35]. HDLs enriched in TGs are hydrolyzed by hepatic lipase, leading to lipid-poor HDLs. Such HDLs are thermodynamically unstable and exhibit structural modifications in apoA-I, which facilitates both its dissociation from HDL particles [40] and its renal glomerular filtration [41].
3. HDL Functions
3.1. Reverse Cholesterol Transport
The best-known function of HDLs is their major role in RCT, which enables the removal of cholesterol from lipid-laden macrophages and artery walls. To sum up, HDLs promote cholesterol efflux from macrophage foam cells in atherosclerotic plaques either specifically by interacting with the transporters ABCA1 and ABCG1, or by aqueous diffusion, through a process facilitated by the scavenger receptor B1 (SR-B1). ABCA1 mediates cholesterol efflux preferentially to lipid-poor apoA-I and small dense HDLs, whereas ABCG1 transports cholesterol to the more mature lipidated HDL particles. Free cholesterol is then esterified by LCAT, and this esterification is important for maintaining the dynamics of cholesterol efflux. Cholesterol is ultimately cleared by the liver, either directly by selective uptake through SR-B1 or by a more recently discovered pathway mediated by F1-ATPase and P2Y13 receptor, or indirectly after CETP-mediated transfer to apoB-containing lipoproteins, which are then internalized by the LDL receptor. In addition to this classical hepatobiliary pathway, cholesterol can be also eliminated by a transintestinal pathway called transintestinal cholesterol efflux (TICE) [42].
Cholesterol efflux is thus the first step in the atheroprotective RCT pathway, and the cholesterol efflux capacity (CEC) of HDL particles is a crucial determinant of cholesterol clearance from lipid-laden macrophages. Over the past few years, studies have demonstrated that the CEC of HDLs is more strongly and inversely associated with incident cardiovascular events than the circulating HDL-C level itself [43][44].
Some authors reported an increased CEC in patients with T2DM [45] and in diabetic patients with hypertriglyceridemia [46]. ABCA1-dependent efflux was also increased using apoB-depleted serum from T2DM patients with hypertriglyceridemia compared to T2DM patients without hypertriglyceridemia [47]. On the other hand, CEC in T2DM patients was unmodified using fibroblasts and whole plasma [46][48][49], human THP-1 macrophages, and apoB-depleted serum [11], or when using THP-1 cells and HDLs isolated by dextran sulfate precipitation [28]. Lastly, CEC was decreased in T2DM patients using adipocytes and LpA-I (i.e., HDL particles containing apoA-I but not apoA-II) [50], Fu5AH hepatoma cells and whole plasma/serum [19][51][52][53][54][55], mouse peritoneal macrophages and isolated HDL3 [56], murine RAW264.7 macrophages and apoB-depleted serum [36], and also THP-1 macrophages and isolated HDLs [1][57]. Similarly, it was recently reported that small HDL particles and apoB-depleted serum from patients with T2DM both have impaired ABCA1-dependent CEC using baby hamster kidney (BHK) cells [36][58]. However, medium and large HDL particles had a similar capacity to promote ABCA1-specific CEC in T2DM patients compared to control individuals in the study [58]. Otherwise, all three sizes of HDL particles from T2DM subjects had similar ABCG1-dependent CEC compared to controls [58].
Changes in lipid composition may also affect HDL CEC. In particular, the replacement of CE by TG molecules in HDLs affects the conformation of apoA-I [15] and could therefore modulate binding to receptors. In addition, the literature suggests that the content of HDLs in total PLs [59][60] and in SMs [61] modulates CEC, but, as mentioned above, the changes in these parameters are very heterogeneous across studies. Cardner et al. recently reported that CEC of apoB-depleted serum is mainly driven by apoA-I level in diabetic individuals [5].
3.2. Anti-Inflammatory Properties
Both diabetes and obesity are associated with low-grade inflammation, which substantially contributes to endothelial dysfunction and atherosclerosis. As shown in Figure 2, HDL particles exert an anti-inflammatory function by downregulating the expression of molecules involved in the recruitment of immune cells into the subendothelial space. These molecules include chemokine CCL-2 [62], vascular cell adhesion molecule (VCAM)-1, intracellular adhesion molecule (ICAM)-1, and selectin-E [11]. In addition, HDLs inhibit the release of inflammatory cytokines, such as TNF-α and IL-1β. The HDL anti-inflammatory function seems of particular relevance for CV outcomes, since it predicts new cardiac events in patients with myocardial infarction, independently of HDL-C [63]. Moreover, an inverse association between the anti-inflammatory capacity of HDLs and incident CV events was recently observed in a study of individuals from the general population cohort, independently of both HDL-C and CEC [64].
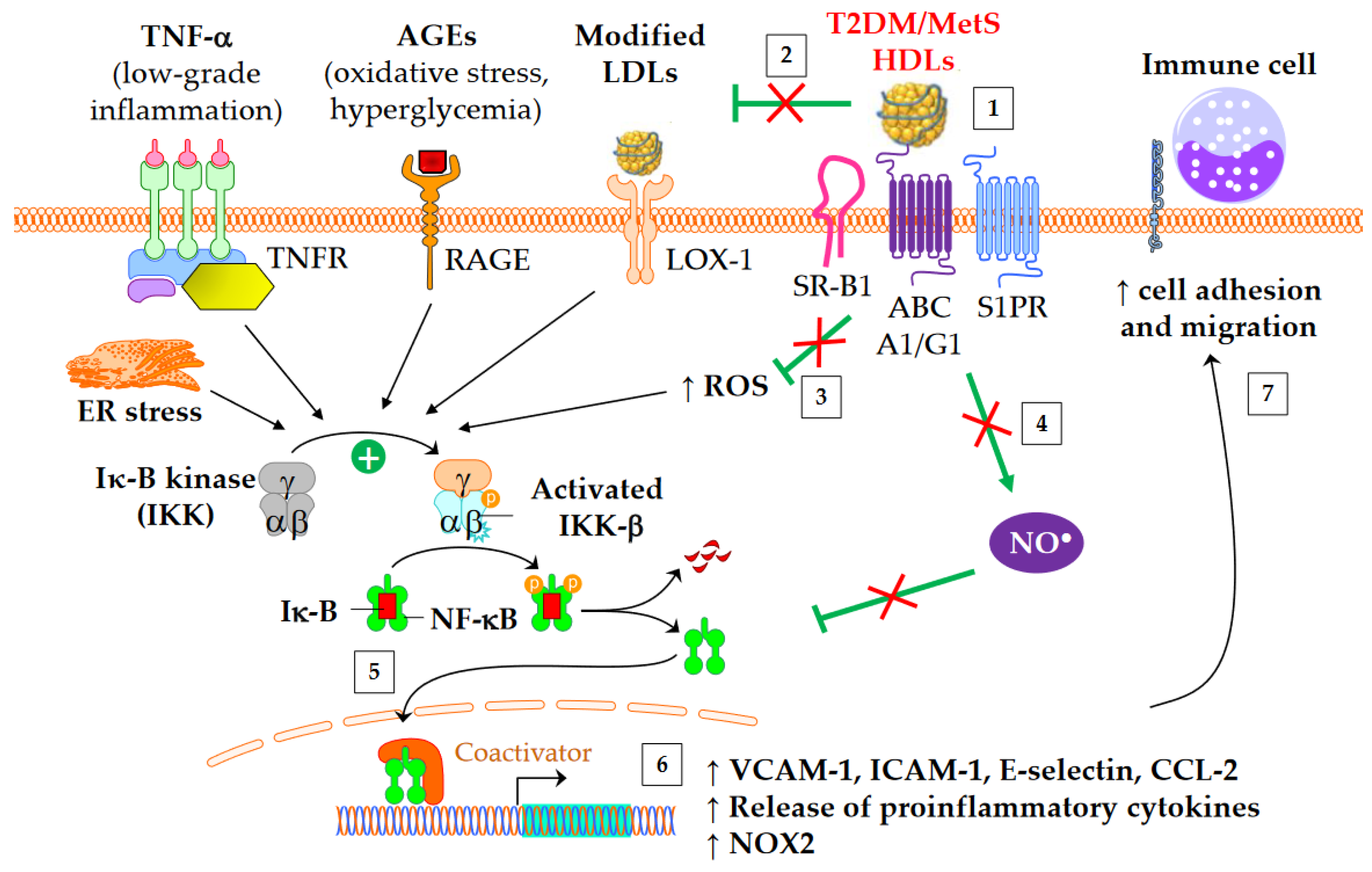
Figure 2. Anti-inflammatory functions of HDLs in T2DM and MetS. ↑ means increase. The inflammatory NF-κB pathway is triggered by several mediators, including TNF-α, advanced glycation end-products (AGEs), modified LDLs, reactive oxygen species (ROS), and endoplasmic reticulum (ER) stress. This leads to an increased gene expression of adhesion molecules, proinflammatory cytokines, and NADPH oxidase (NOX2). Green arrows represent the functions of healthy HDLs. (1) The binding of HDLs to receptors is modified by conformational changes in HDL particles in insulin resistant conditions. The depletion in S1P of MetS HDLs likely decreases the binding to S1P receptors (S1PR). (2) HDL-mediated protection of LDLs against oxidation is affected, (3) in particular due to the loss of capacity of HDLs to dampen ROS production. This promotes the NF-κB activation triggered by the recognition of oxidized LDLs by scavenger receptors in vasculature, particularly by LOX-1 (i.e., SR-E1) and SR-A1. (4) The activation of endothelial NO synthase by HDLs is reduced (see 4.3) and subsequently inhibits nitrosylation of NF-κB. (5) Ultimately, HDLs are less able to inhibit the translocation of NF-κB into the nucleus, and, (6) afterwards, the gene expression of adhesion molecules and proinflammatory cytokines. (7) This facilitates the recruitment of immune cells into the subendothelial space.
HDLs from T2DM patients are less able to inhibit the migration of monocytes towards endothelial cells [32][65]. Interestingly, this loss of function correlates with plasma SAA [65] and carbamylated HDL levels [32]. The fact that in vitro carbamylation of HDLs reproduces the loss of capacity to inhibit the migration of monocytes reinforces the potential role of carbamylated HDLs [32]. Although HbA1c or glucose levels do not correlate with this loss of HDL function [65], some evidence suggests that glycoxidative changes in HDLs may play a role. Thus, the ex vivo treatment of plasma with L-4F, an apoA-I mimetic peptide able to bind oxidized lipids with a higher affinity than apoA-I itself [66], restores the anti-inflammatory function of HDLs [65].
The activation of the classical IKK/IκB-α/NF-κB pathway plays a crucial role in the expression of adhesion molecules and in the release of proinflammatory cytokines into cardiovascular tissue. HDLs/apoA-I inhibit the NF-κB pathway via several overlapped mechanisms following interaction with SR-B1, S1PR, ABCA1, and ABCG1: cholesterol efflux, endothelial nitric oxide synthase (eNOS) activation [67] and subsequent S-nitrosylation [68], upregulation of 3β-hydroxysteroid-δ24 reductase [69][70] and heme oxygenase-1 [70], and inhibition of the toll-like receptors TLR4 [71] and TLR2 [72]. HDLs or apoA-I ultimately prevent NF-κB p65 subunit translocation to the nucleus and DNA binding [71], thus inhibiting the transcription of adhesion molecules, CCL-2, proinflammatory cytokines, and also NADPH-oxidase (NOX2) genes. It has been shown that HDLs isolated from T2DM patients are unable to suppress the activating phosphorylation of the NF-κB p65 subunit in endothelial cells [73]. Glycated apoA-I partially loses its ability to inhibit cytosolic IκB-α phosphorylation and NF-κB p65 subunit translocation to the nucleus [22].
3.3. Antioxidative Properties
T2DM and obesity are known to be associated with increased oxidative stress, which is closely linked to low-grade inflammation. In particular, oxidative stress in artery walls is responsible for the formation of oxidized LDLs (oxLDLs), rendering them more atherogenic. There are several mechanisms through which HDLs present in the intima can protect LDLs against oxidation [74]. HDLs directly protect LDLs from oxidation induced by one-electron oxidants (free radicals), and they are also able to remove oxidized lipids from LDLs. These activities can decrease local concentrations of oxLDLs. The antioxidative potential of HDL particles originates both from the activities of their proteins and from lipid components. Different HDL-associated apolipoproteins, lipid transfer proteins, and enzymes have been shown to contribute to the antioxidative capacity of HDLs.
3.4. Nitric Oxide Production
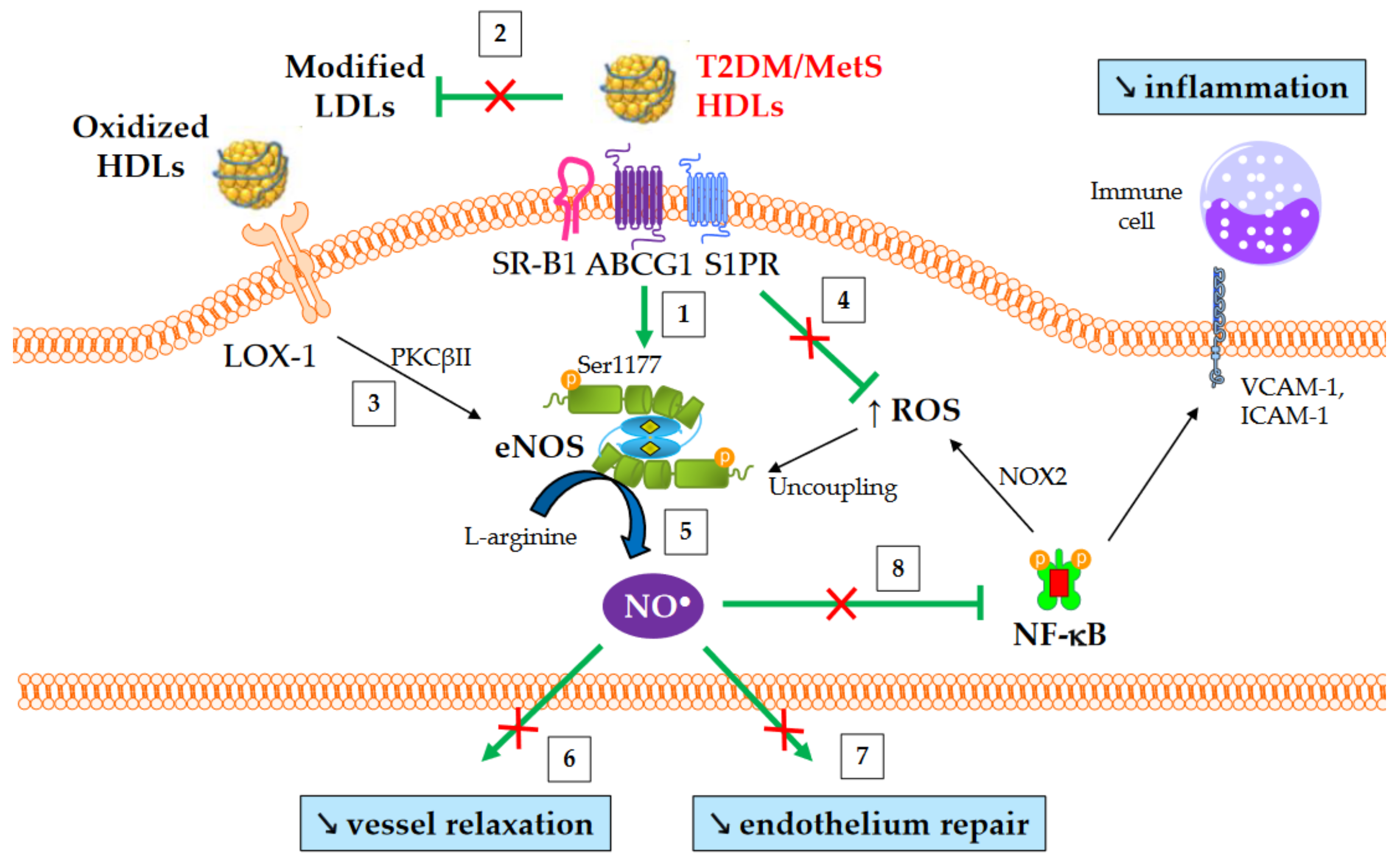
The production of nitric oxide (NO) is important for normal endothelial function and protects against endothelial dysfunction, an early hallmark of atherosclerosis. HDLs improve the bioavailability of NO in vasculature mainly by inducing the activating-phosphorylation of eNOS at serine 1177, which then promotes NO synthesis. NO production contributes to a number HDL’s beneficial effects, such as vasorelaxation, inhibition of NF-κB pathway, and endothelium repair [75]. Several mechanisms are involved in eNOS activation mediated by HDLs. These include the binding of HDL-apoA-I to SR-B1 [76] and ABCG1 [77], the binding of HDL-S1P to S1PR1/3 receptors [78], the activation of mitogen-activated protein (MAP) kinases [79], the inhibition of protein kinase C (PKC) βII [67], cholesterol efflux facilitating the dissociation of eNOS from caveolin-1 [77], and, lastly, the suppression of ROS production which preserves from eNOS uncoupling.
An overview of NO-mediated HDL functions in T2DM and MetS is presented in Figure 3. HDLs from T2DM patients have been demonstrated to be less able to induce the activating-phosphorylation of eNOS at serine 1177 [73], NO production [27], and vessel relaxation [27]. Interestingly, eNOS phosphorylation and activity are even affected in obese patients in the absence of diabetes [80].
Figure 3. NO-mediated HDL functions in T2DM and MetS. ↑ and ↘ mean increase and decrease, respectively. Green arrows represent the functions of healthy HDLs. (1) HDLs from T2DM and non-diabetic MetS individuals are less able to induce Akt-dependent eNOS phosphorylation at Ser1177. (2) HDL-mediated protection of LDLs against oxidation is affected, thus facilitating eNOS inhibition after the binding of modified LDLs to LOX-1. (3) Oxidized HDLs are also able to bind to LOX-1 receptor, leading to PKCβII activation and subsequently to eNOS inhibition. (4) The loss of capacity of HDLs to dampen ROS production promotes eNOS uncoupling. (5) Ultimately, HDL-mediated NO production is reduced, (6) affecting the relaxation of vascular smooth muscle cells and (7) endothelium repair. (8) Reduced NO synthesis diminishes the inhibition of nitrosylation of NF-κB, as well as NOX2-mediated ROS production and the recruitment of immune cells into the subendothelial space.
3.5. Antiapoptotic Properties and Endothelium Repair
The integrity of endothelial cells is crucial for vascular homeostasis, and endothelial cell death triggers vascular damage and promotes inflammation and endothelial dysfunction. HDL particles can inhibit apoptosis in endothelial cells, thus preserving endothelium integrity [81][82][83].
The antiapoptotic activity of small HDL particles is reduced in MetS individuals [84][85], and is closely associated with altered physicochemical properties, such as core CE depletion and TG enrichment in apoA-I-poor HDL3c [84].
The depletion of HDLs in plasmalogens in T2DM and MetS may also play a role, since enrichment of rHDLs with plasmalogens enhances their antiapoptotic activity [81]. Changes in the HDL-apoM/S1P axis are also likely to be involved considering its important role in HDL antiapoptotic activity [82][86].
4. Antidiabetic Properties
Emerging data suggest that HDL particles could actually contribute to the development of diabetes. Firstly, HDL-C levels are inversely associated with T2DM development in epidemiological studies [87][88][89], and this metric has been included in scores for T2DM risk [89][90]. In addition, Mendelian randomization studies showed that HDL-C elevation is associated with a lower risk of developing T2DM [91][92]. Moreover, both HDL particle size and the concentration of large HDL particles were inversely associated with incident T2DM in the general population [4]. Moreover, CETP inhibitors, which increased HDL-C concentration of 29 to 132% in large interventional studies, reduced the risk of new-onset diabetes by 16% on average [93].
Many of the antidiabetic mechanisms induced by HDLs or apoA-I have now been identified [94]. Infusions of rHDLs and apoA-I stimulate insulin secretion and reduce plasma glucose concentrations in obese mice and in T2DM patients [95][96]. From a mechanistic point of view, apoA-I enhances the expression of key enzymes involved in insulin maturation in β-cells [97]. In addition, HDLs protect β-cells from apoptosis induced by endoplasmic reticulum stressors [98].