Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Theoretical examination of the antioxidant activity of the individual or selected compounds identified in a matrix can be a green mid-tool for their prioritization regarding antioxidant behavior before seeking experimental in vitro or in vivo data.
- antioxidant activity
- theoretical predictions
- phenolic compounds
- olive tree products
- olive tree by-products
- oleuropein
- hydroxytyrosol
- tyrosol
- secoiridoids
- density functional theory
1. Introduction
The interest in natural compounds that exhibit antioxidant activity in food matrices [1] is extended also to the benefits such compounds can exert on humans or animal species after consumption [2][3]. For example, the high oxidative stability of virgin olive oil (VOO) at first was attributed to its rich in monounsaturated fatty acids character and then to the critical role of certain groups of intrinsic antioxidants such as the phenolic compounds present in the polar fraction of the oil. For more than two decades, the interest has been focused on the in vivo properties of the secoiridoids oleuropein, ligstroside, and related derivatives, which can be found not only in the oil but also in other edible products of the olive tree or can be isolated from by-products of the olive industry [4][5].
Antioxidants, in general, exhibit their activity even when present in minute concentrations. The phenolic-type antioxidants are considered primary antioxidants because they react directly with reactive oxygen and nitrogen species via hydrogen atom and/or electron donation and have gained the interest of food, medical, pharmaceutical, and cosmeceutical researchers all over the world [6].
2. Theoretical Prediction Strategies
As a result of advances in computer science and software engineering, it is feasible to accomplish theoretical calculations for compounds of medium or even larger size in a reasonable amount of time, making them suitable even for routine studies. The overall number of publications regarding antioxidants that involve theoretical calculations is steadily increasing, but the latter have not found their place in the agriculture and food sector yet. This is indicated by the bibliometrics shown in Figure 1.
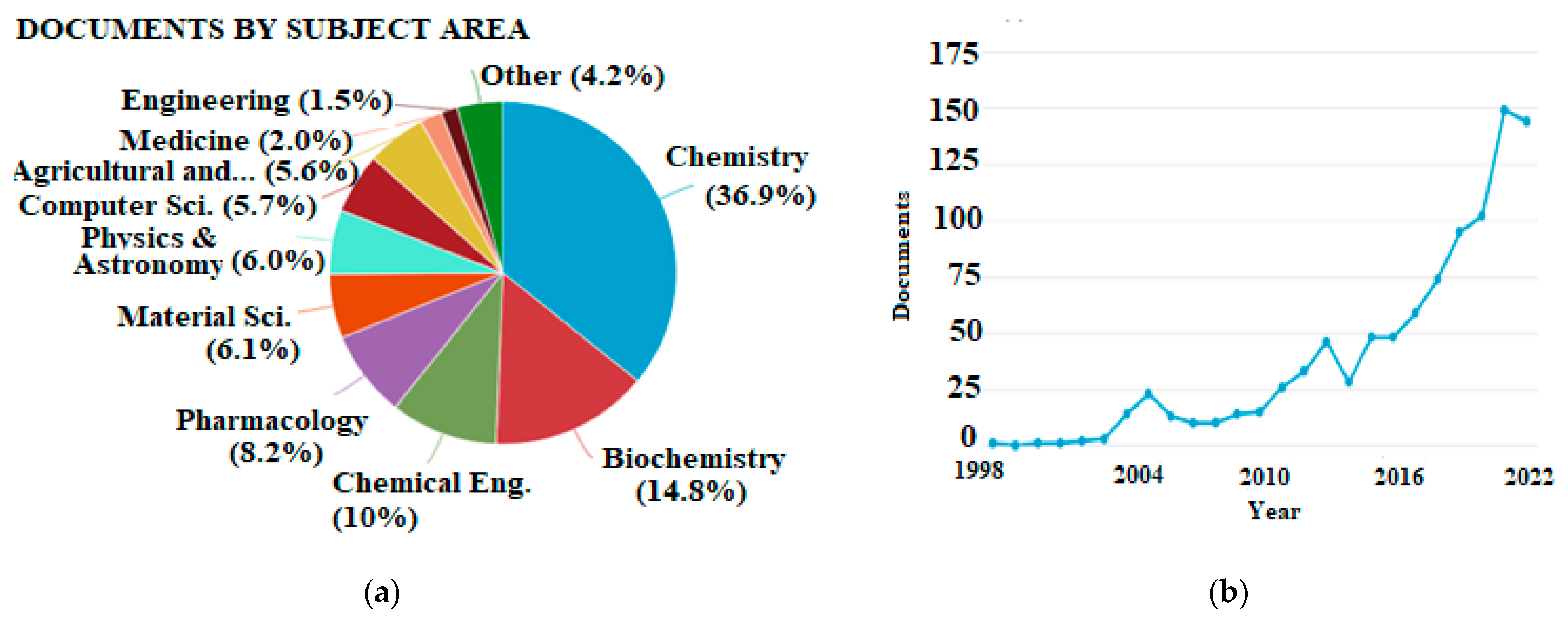
Figure 1. (a) Publications categorized by subject area and (b) total publications per year appearing in Scopus database using “density functional theory” and “antioxidant” as keywords (accessed 21 November 2022, total documents, 995).
In most of the relevant publications, emphasis is given to the study of the radical scavenging process that coincides with the major mechanisms of action of primary antioxidants, such as the phenolic compounds [7][8][9][10][11][12][13][14].
Different strategies can be applied to theoretical studies so that the derived data are reported differently, for example, in terms of intrinsic reactivity indices, or using thermochemical or kinetic parameters. In all cases, knowledge of the possible reactions involved and the factors that may influence the reactivity (e.g., environment) is a prerequisite [15].
The theoretical approach usually begins with the conformer analysis to locate the most stable compound (global minimum). This requires the examination of all possible structures, often including the determination of enthalpy barriers upon, e.g., rotation of one or more dihedral angles. More elaborate is the potential energy surface (PES) examination, which results in a plot of the energy versus geometry with concomitant rotation of dihedrals. Such an approach can be useful for equilibrium geometries determination and transition states [16][17]. An example of targeted PES examination with a concomitant rotation of two dihedrals is given in Figure 2 for the ligstroside derivative tyrosol. The contour map indicates the place of global minimum, local minima, and destabilization.
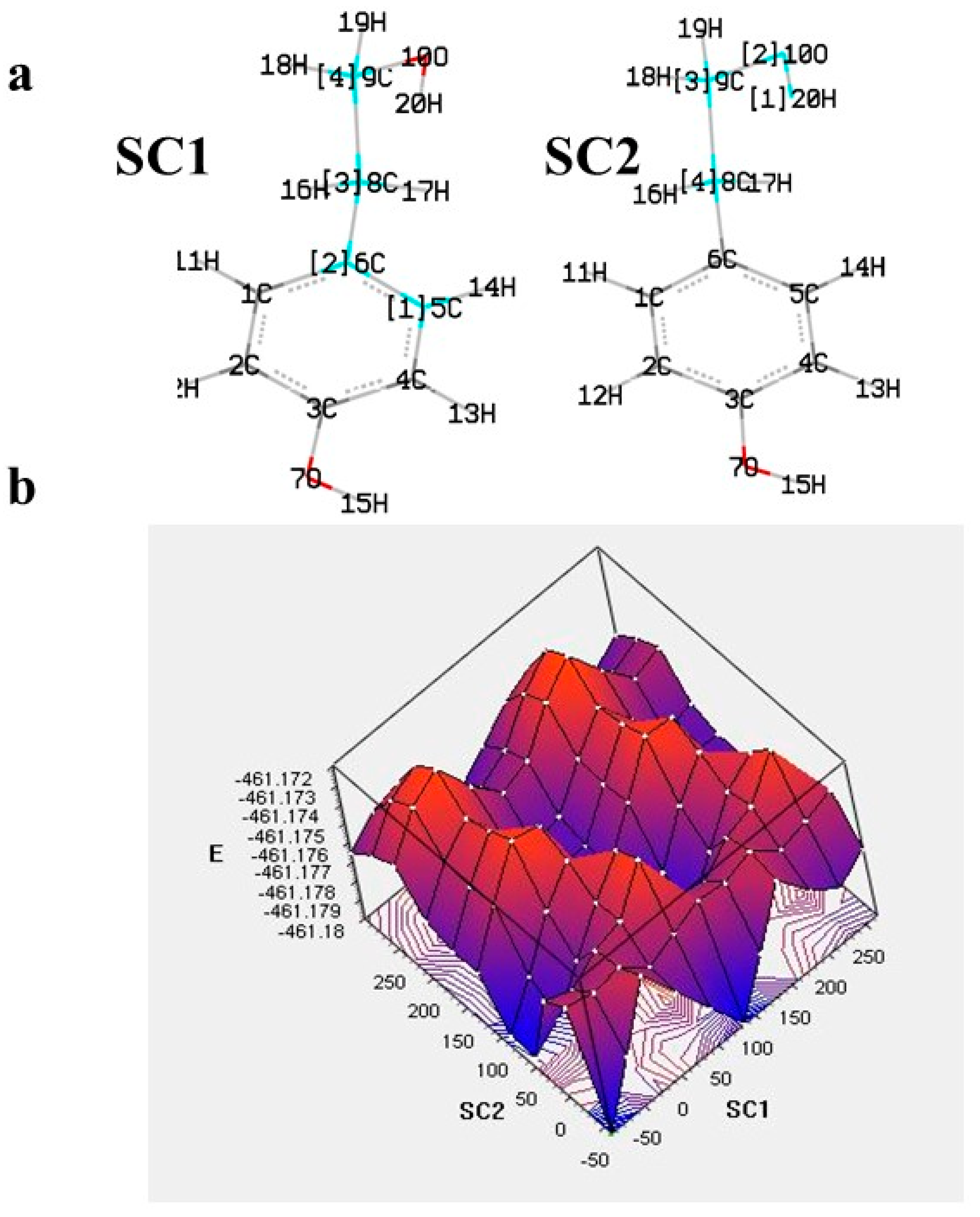
Figure 2. (a) The varying dihedrals of tyrosol (SC1: C5-C6-C8-C9, SC2: C8-C9-O10-H20) selected for the potential energy surface (PES) scan at B3LYP/6-31G in gas phase and (b) 3D plot of PES [18].
Certainly, it is possible to locate more than one conformer, in case the difference in enthalpy from the global minimum is small, suggesting that interconversion can be feasible. Except for the rotation of groups and side chains, tautomerism and shifting from E to Z configuration or bending of dihedral angles are features to be also explored, as highlighted by Spiegel [7]. All of them can affect hydrogen bonding, electron delocalization, polarizability, dipole moment value, as well as steric hindrance phenomena.
Considering that an optimum structure has been obtained, then a first-cut approach resides in the frontier orbital examination. The highest molecular orbital (HOMO) energy is related to electron donation, and the lowest unoccupied molecular orbital (LUMO) to electron acceptance [7][19]. Thus, the energy gap between the two orbitals, electronegativity (χ), chemical potential (μ), global hardness (η), global softness (S), and electrophilicity (ω) can be calculated to have an estimate of molecular reactivity. However, electron correlation (a measure of the influence in an electron movement due to the presence of all other electrons in the system), which is critical for accurate and quantitative evaluation of molecular energies, is not considered and may affect the validity of conclusions [19]. In addition, visualization of HOMO may serve in detecting the structural site of the attack, even though the electrophilic/nucleophilic sites are better revealed via solving the Fukui functionals [20] and more accurately by applying the dual descriptor proposed by Martínez-Araya [21].
Another approach is the examination of the so-called intrinsic reactivity of the antioxidants, which is most frequently employed by different research groups [15]. Such an approach requires the calculation of enthalpy of derived species, namely phenoxy radical, cation radical, anion, hydrogen atom, and proton, even of further derivatives depending on whether the structure favors more than one proton or electron donation in a stepwise manner (e.g., double-HAT, double-SPLET, SPLHAT) [14], which in turn are used to calculate the indices given in Table 1.
Table 1. Common molecular indices related to the radical scavenging potential of compounds and the formulae for their calculation.
| Molecular Index | Formula * |
|---|---|
| Bond dissociation enthalpy (BDE) | Hr + Hh − Hp |
| Adiabatic ionization potential (IP) | Ecr − Ep |
| Proton dissociation enthalpy (PDE) | Hr + Hpr − Hcr |
| Proton affinity (PA) | Ha + Hpr − Hp |
| Electron transfer energy (ETE) ** | Er − Ea |
Where * Hr: enthalpy of phenoxy radical, Hh: enthalpy of the H-atom, Hp: enthalpy of the parent molecule, Ecr: energy of the cation radical, Ep: energy of the parent molecule, Hpr: enthalpy of the proton, Hcr: enthalpy of the cation radical, Ha: enthalpy of the anion, Er: energy of the phenoxy radical, Ea: energy of the anion. (** In some publications, the enthalpies are considered, including also in the formula the enthalpy of the electron).
In addition to the above, in some studies, to comment on the reactivity and the dominant mechanism, the radical species of interest are also considered, and their BDE and electron affinity (EA) values are computed as well [22]. As the effectiveness of a radical scavenger is not only related to the ease of hydrogen atom and/or electron donation, but also to the stability of the phenoxy radical formed through intramolecular bonding or extension of conjugation, the spin distribution can be obtained and, particularly, the value of the localized spin in the oxygen of the corresponding radical, which can be a useful index of radical stability. The latter is illustrated in Figure 3 for the two phenoxy radicals of oleuropein [23]. Due to the lack of extended conjugation, the electron spin is delocalized only in the aromatic ring. Furthermore, the radical formed from the p-OH group is more stable than that from the m-OH group, as is evident by the spin value in the oxygen of the corresponding radicals (0.35 vs. 0.39).
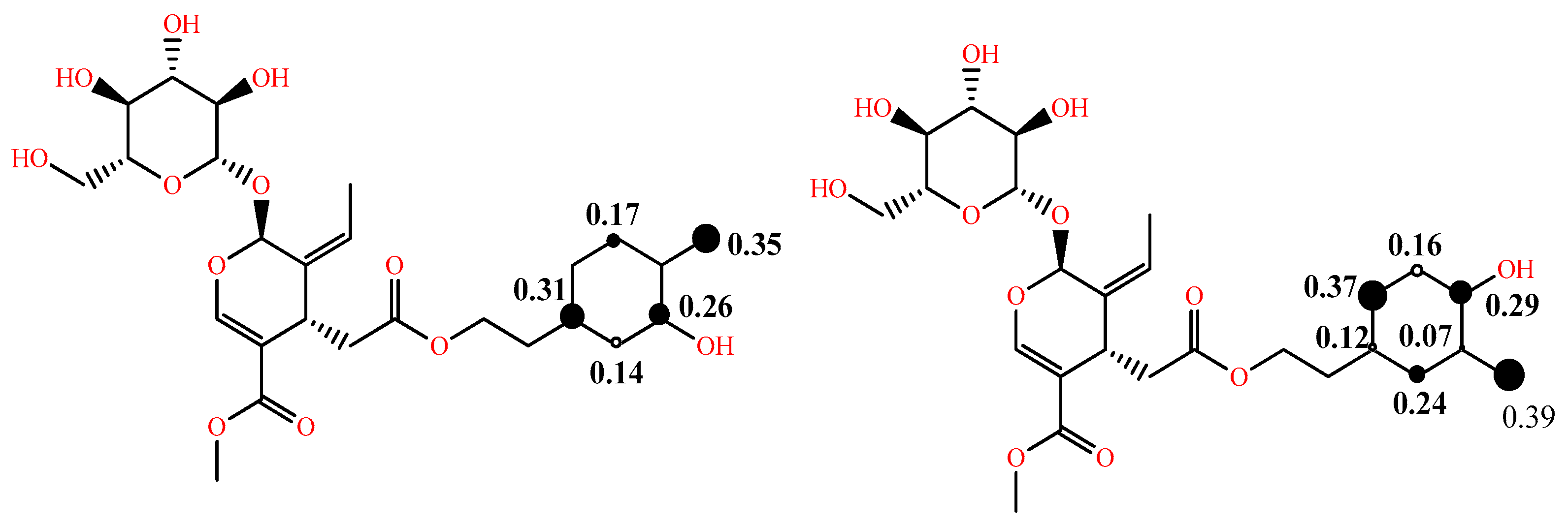
Figure 3. Spin density distribution in phenoxy radicals of oleuropein in the gas phase at B3LYP/6-31G (d,p) level of theory adapted from [23] supplementary; larger size of the black dot corresponds to higher spin density.
When selecting to work from a thermochemical point of view, the calculation of the Gibbs free energy (ΔG) value is the most appropriate index to examine a specific reaction pathway. According to the equation ΔG = ΔH − Δ(TS), its calculation, except for temperature (T), also includes the entropy (S) of the system that is influenced by the mechanism governing the reaction. The approach is considered to offer more valuable information compared to that obtained from the calculation of indices of intrinsic reactivity, as both the reactivity of the antioxidant and that of a particular free radical species are co-evaluated. The latter is important, considering that the various free radicals to be scavenged differ in reactivity. A drawback, as highlighted by Galano and Alvarez-Idaboy [15], is that when using such a strategy, kinetics is not taken into consideration, and in some cases, opposite trends may be observed due to deviation from pertinence to the Bell–Evans–Polanyi principle, i.e., the usual linear relationship between the activation energy and enthalpy of reaction. Furthermore, it should be stressed that to make feasible the calculation, small-size radicals are usually selected, such as HO●, O2●−, CH3OO•, and HOO• [24][25].
When a kinetic study is designed for a biological antioxidant, the starting point of consideration is that, according to the definition of Halliwell et al. [26], a compound should react faster with the oxidizing species than the substrate to be considered as a potentially efficient antioxidant. The advantages of kinetic approaches, according to Galano and Alvarez-Idaboy [15], are the following:
-
Consideration of tunneling effects for reactions involving small particles;
-
Contribution of different mechanisms of reaction, and reaction sites, to the whole activity of the examined compound;
-
Inclusion of reactions that do not fulfill the Bell–Evans–Polanyi principle;
-
Incorporation of the pH influence on the reactivity of antioxidants bearing ionizable groups such as acids, considering the molar fraction of different forms present when the pH value is different;
-
Consideration of single electron transfer reactions situated in the inverted region of the Marcus parabola.
In a kinetic study, except for the overall rate constant of reaction, the contribution of different mechanisms can be estimated via calculation of the corresponding rates to eventually obtain the so-called branching ratios (Γ) according to the following formula:
,
where ki represents the rate constant of the mechanism or pathway of interest. A typical example is given below for hydroxytyrosol (Table 2), indicating that the main mechanism involved in the scavenging is the hydrogen atom transfer by ~98% regardless of the polarity of the media. In the lipid media, hydrogen atom transfer from the 1a position was found to be the most preferable, whereas in water, the hydrogen transfer was equally feasible from 1a and 1b due to diffusion limitation [27].
Table 2. Branching ratios (Γ) for the most important mechanisms and pathways involving hydroxytyrosol and methoxy radical in pentyl ethanoate and water (data abstracted from Table 2 of cited reference [27] and adjusted properly).
| Structure and Numbering | Pentyl Ethanoate | Water |
|---|---|---|
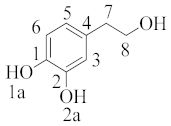 hydroxytyrosol |
HAT(1a) = 61.53 | HAT(1a) = 49.69 |
| HAT(2a) = 37.69 | HAT(2a) = 49.69 | |
| HAT(7) = 0.06 | HAT(7) = 0.03 | |
| HAT(8) = 0.14 | HAT(8) = 0.10 | |
| RAF*(1) = 0.55 | RAF(1) = 0.20 | |
| RAF(2) = 0.00 | RAF(2) = 0.12 | |
| RAF(3) = 0.00 | RAF(3) = 0.01 | |
| RAF(4) = 0.03 | RAF(4) = 0.16 | |
| RAF(5) = 0.00 | RAF(5) = 0.01 | |
| RAF(6) = 0.00 | RAF(6) = 0.00 |
* RAF = radical adduct formation.
This entry is adapted from the peer-reviewed paper 10.3390/molecules28052267
References
- Brewer, M. Natural antioxidants: Sources, compounds, mechanisms of action, and potential applications. Compr. Rev. Food Sci. Food Saf. 2011, 10, 221–247.
- Hrelia, S.; Angeloni, C. New mechanisms of action of natural antioxidants in health and disease. Antioxidants 2020, 9, 344.
- Salami, S.A.; Guinguina, A.; Agboola, J.O.; Omede, A.A.; Agbonlahor, E.M.; Tayyab, U. Review: In vivo and postmortem effects of feed antioxidants in livestock: A review of the implications on authorization of antioxidant feed additives. Animal 2016, 10, 1375–1390.
- Boskou, D.; Tsimidou, M.; Blekas, G. Polar phenolic compounds. In Olive Oil: Chemistry and Technology, 2nd ed.; Boskou, D., Ed.; AOCS Press: Champaign, IL, USA, 2006; pp. 73–92.
- Nenadis, N.; Papoti, V.T.; Tsimidou, M.Z. Bioactive ingredients in olive leaves. In Olives and Olive Oil in Health and Disease Prevention, 2nd ed.; Preedy, V.R., Watson, R.R., Eds.; Academic Press: San Diego, CA, USA, 2021; pp. 65–78.
- Zeb, A. Concept, mechanism, and applications of phenolic antioxidants in foods. J. Food Biochem. 2020, 44, e13394.
- Spiegel, M. Current trends in computational quantum chemistry studies on antioxidant radical scavenging activity. J. Chem. Inf. Model. 2022, 62, 2639–2658.
- Tsimidou, M.; Nenadis, N.; Zhang, H.Y. Structure radical scavenging activity relationships of flavonoids and phenolic acids. In Natural Antioxidant Phenols. Sources, Structure—Activity Relationship, Current Trends in Analysis and Characterization; Boskou, D., Gerothanassis, I., Kefalas, P., Eds.; Research Signpost: Kerala, India, 2006; pp. 33–51.
- Nenadis, N.; Tsimidou, M.Z. On the use of DFT computations to the radical scavenging activity studies of natural phenolic compounds. In Density Functional Theory: Principles, Applications and Analysis; Morin, J., Pelletier, J.M., Eds.; Nova Science Publishers Inc.: New York, NY, USA, 2013; pp. 121–146.
- Leopoldini, M.; Russo, N.; Toscano, M. The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem. 2011, 125, 288–306.
- Alov, P.; Tsakovska, I.; Pajeva, I. Computational studies of free radical-scavenging properties of phenolic compounds. CTMC 2015, 15, 85–104.
- Galano, A.; Mazzone, G.; Alvarez-Diduk, R.; Marino, T.; Alvarez-Idaboy, J.R.; Russo, N. Food antioxidants: Chemical insights at the molecular level. Annu. Rev. Food Sci. Technol. 2016, 7, 335–352.
- Mahmoudi, S.; Dehkordi, M.M.; Asgarshamsi, M.H. Density functional theory studies of the antioxidants—A Review. J. Mol. Model. 2021, 27, 271.
- Mittal, A.; Vashistha, V.K.; Das, D.K. Recent advances in the antioxidant activity and mechanisms of chalcone derivatives: A computational review. Free Radic. Res. 2022, 56, 378–397.
- Galano, A.; Raúl Alvarez-Idaboy, J. Computational strategies for predicting free radical scavengers’ protection against oxidative stress: Where are we and what might follow? Int. J. Quantum Chem. 2019, 119, e25665.
- Lewars, E.G. The concept of the potential energy surface. In Computational Chemistry; Lewars, E.G., Ed.; Springer: Cham, Switzerland, 2016; pp. 9–49.
- Semidalas, C.; Semidalas, E.; Matsoukas, M.T.; Nixarlidis, C.; Zoumpoulakis, P. In silico studies reveal the mechanisms behind the antioxidant and anti-inflammatory activities of hydroxytyrosol. Med. Chem. Res. 2016, 25, 2498–2511.
- Nenadis, N.; (Aristotle University of Thessaloniki, School of Chemistry, Thessaloniki, Greece); Pyrka, I.; (Aristotle University of Thessaloniki, School of Chemistry, Thessaloniki, Greece); Tsimidou, M.Z.; (Aristotle University of Thessaloniki, School of Chemistry, Thessaloniki, Greece). Unpublished work, 2023.
- Domingo, L.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748.
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050.
- Martínez-Araya, J.I. Why Is the dual descriptor a more accurate local reactivity descriptor than Fukui functions? J. Math. Chem. 2015, 53, 451–465.
- Wang, L.-F.; Zhang, H.-Y. A theoretical investigation on DPPH radical-scavenging mechanism of edaravone. Bioorg. Med. Chem. Lett. 2003, 13, 3789–3792.
- Hassanzadeh, K.; Akhtari, K.; Hassanzadeh, H.; Zarei, S.A.; Fakhraei, N.; Hassanzadeh, K. The role of structural C–H compared with phenolic OH sites on the antioxidant activity of oleuropein and its derivatives as a great non-flavonoid family of the olive components: A DFT study. Food Chem. 2014, 164, 251–258.
- Amić, A.; Mastiľák Cagardová, D. DFT study of the direct radical scavenging potency of two natural catecholic compounds. Int. J. Mol. Sci. 2022, 23, 14497.
- Lu, Y.; Wang, A.; Shi, P.; Zhang, H.; Li, Z. Quantum chemical study on the antioxidation mechanism of piceatannol and isorhapontigenin toward hydroxyl and hydroperoxyl radicals. PLoS ONE 2015, 10, e0133259.
- Halliwell, B.; Aeschbach, R.; Löliger, J.; Aruoma, O.I. The characterization of antioxidants. Food Chem. Toxicol. 1995, 33, 601–617.
- Galano, A.; Alvarez-Idaboy, J.R.; Francisco-Márquez, M.; Medina, M.E. A Quantum chemical study on the free radical scavenging activity of tyrosol and hydroxytyrosol. Theor. Chem. Acc. 2012, 131, 1173.
This entry is offline, you can click here to edit this entry!