DNA repair pathways are essential for ensuring normal DNA synthesis, genomic stability, and integrity, which are required for a multitude of cellular processes such as cell proliferation, differentiation, cell cycle, apoptosis, and the development of tissues and organs. Defects or inappropriateness in DNA repair pathways are associated with detrimental health effects, including birth defects, cancer, and neurodegenerative diseases. DNA repair genes are over-expressed at the early stages of normal embryonic development in order to reduce possible replication errors and genotoxic damage. The occurrence of NTDs may be related to the abnormality or deletion of various DNA repair pathways. Due to the link between folate deficiency and NER not being particularly evident, we mainly review the progress of DNA repair pathways (BER, MMR, DSBR) and NTDs with folate deficiency.
- neural tube defects
- DNA damage
- DNA repair
- folate
1. MMR and NTDs
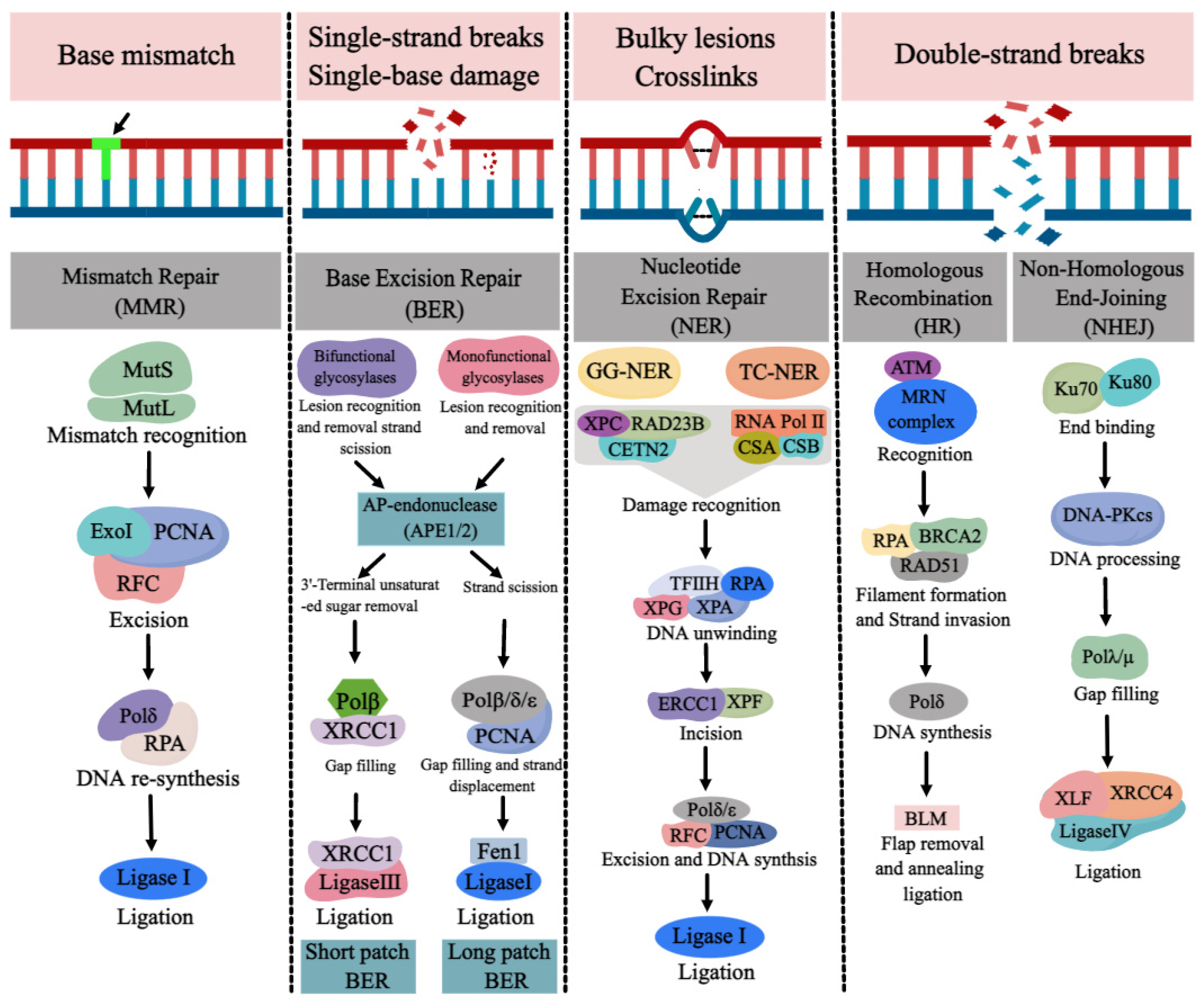
2. BER and NTDs
3. DSBR and NTDs
This entry is adapted from the peer-reviewed paper 10.3390/ijms24032220
References
- Fukui, K.; Baba, S.; Kumasaka, T.; Yano, T. Structural Features and Functional Dependency on beta-Clamp Define Distinct Subfamilies of Bacterial Mismatch Repair Endonuclease MutL. J. Biol. Chem. 2016, 291, 16990–17000.
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313.
- Baretti, M.; Le, D.T. DNA mismatch repair in cancer. Pharmacol. Ther. 2018, 189, 45–62.
- Niv, Y. Microsatellite instability and MLH1 promoter hypermethylation in colorectal cancer. World J. Gastroenterol. 2007, 13, 1767–1769.
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3.
- Cravo, M.L.; Albuquerque, C.M.; Salazar de Sousa, L.; Gloria, L.M.; Chaves, P.; Dias Pereira, A.; Nobre Leitao, C.; Quina, M.G.; Costa Mira, F. Microsatellite instability in non-neoplastic mucosa of patients with ulcerative colitis: Effect of folate supplementation. Am. J. Gastroenterol. 1998, 93, 2060–2064.
- Liu, Z.; Wang, Z.; Li, Y.; Ouyang, S.; Chang, H.; Zhang, T.; Zheng, X.; Wu, J. Association of genomic instability, and the methylation status of imprinted genes and mismatch-repair genes, with neural tube defects. Eur. J. Hum. Genet. 2012, 20, 516–520.
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013, 153, 590–600.
- Hu, M.; Sun, X.J.; Zhang, Y.L.; Kuang, Y.; Hu, C.Q.; Wu, W.L.; Shen, S.H.; Du, T.T.; Li, H.; He, F.; et al. Histone H3 lysine 36 methyltransferase Hypb/Setd2 is required for embryonic vascular remodeling. Proc. Natl. Acad. Sci. USA 2010, 107, 2956–2961.
- Li, H.; Wang, X.; Zhao, H.; Wang, F.; Bao, Y.; Guo, J.; Chang, S.; Wu, L.; Cheng, H.; Chen, S.; et al. Low folate concentration impacts mismatch repair deficiency in neural tube defects. Epigenomics 2020, 12, 5–18.
- Riccio, A.; Aaltonen, L.A.; Godwin, A.K.; Loukola, A.; Percesepe, A.; Salovaara, R.; Masciullo, V.; Genuardi, M.; Paravatou-Petsotas, M.; Bassi, D.E.; et al. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat. Genet. 1999, 23, 266–268.
- Pinto, M.; Wu, Y.; Suriano, G.; Mensink, R.G.; Duval, A.; Oliveira, C.; Carvalho, B.; Hamelin, R.; Seruca, R.; Hofstra, R.M. MBD4 mutations are rare in gastric carcinomas with microsatellite instability. Cancer Genet. Cytogenet. 2003, 145, 103–107.
- Bellacosa, A. Role of MED1 (MBD4) Gene in DNA repair and human cancer. J. Cell. Physiol. 2001, 187, 137–144.
- Cortellino, S.; Wang, C.; Wang, B.; Bassi, M.R.; Caretti, E.; Champeval, D.; Calmont, A.; Jarnik, M.; Burch, J.; Zaret, K.S.; et al. Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev. Biol. 2009, 325, 225–237.
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583.
- Kim, Y.J.; Wilson, D.M., 3rd. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13.
- Grundy, G.J.; Parsons, J.L. Base excision repair and its implications to cancer therapy. Essays Biochem. 2020, 64, 831–843.
- Nemec, A.A.; Wallace, S.S.; Sweasy, J.B. Variant base excision repair proteins: Contributors to genomic instability. Semin. Cancer Biol. 2010, 20, 320–328.
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220.
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507.
- Woodhouse, B.C.; Dianova, I.I.; Parsons, J.L.; Dianov, G.L. Poly(ADP-ribose) polymerase-1 modulates DNA repair capacity and prevents formation of DNA double strand breaks. DNA Repair 2008, 7, 932–940.
- Matsumoto, Y.; Kim, K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 1995, 269, 699–702.
- Odell, I.D.; Barbour, J.E.; Murphy, D.L.; Della-Maria, J.A.; Sweasy, J.B.; Tomkinson, A.E.; Wallace, S.S.; Pederson, D.S. Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol. Cell. Biol. 2011, 31, 4623–4632.
- Cappelli, E.; Taylor, R.; Cevasco, M.; Abbondandolo, A.; Caldecott, K.; Frosina, G. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J. Biol. Chem. 1997, 272, 23970–23975.
- Frosina, G.; Fortini, P.; Rossi, O.; Carrozzino, F.; Raspaglio, G.; Cox, L.S.; Lane, D.P.; Abbondandolo, A.; Dogliotti, E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996, 271, 9573–9578.
- Podlutsky, A.J.; Dianova, I.I.; Podust, V.N.; Bohr, V.A.; Dianov, G.L. Human DNA polymerase beta initiates DNA synthesis during long-patch repair of reduced AP sites in DNA. EMBO J. 2001, 20, 1477–1482.
- Bianchi, F.T.; Berto, G.E.; Di Cunto, F. Impact of DNA repair and stability defects on cortical development. Cell. Mol. Life Sci. 2018, 75, 3963–3976.
- Cabelof, D.C.; Raffoul, J.J.; Nakamura, J.; Kapoor, D.; Abdalla, H.; Heydari, A.R. Imbalanced base excision repair in response to folate deficiency is accelerated by polymerase beta haploinsufficiency. J. Biol. Chem. 2004, 279, 36504–36513.
- Unnikrishnan, A.; Prychitko, T.M.; Patel, H.V.; Chowdhury, M.E.; Pilling, A.B.; Ventrella-Lucente, L.F.; Papakonstantinou, E.V.; Cabelof, D.C.; Heydari, A.R. Folate deficiency regulates expression of DNA polymerase beta in response to oxidative stress. Free Radic. Biol. Med. 2011, 50, 270–280.
- Duthie, S.J.; Hawdon, A. DNA instability (strand breakage, uracil misincorporation, and defective repair) is increased by folic acid depletion in human lymphocytes in vitro. FASEB J. 1998, 12, 1491–1497.
- Wang, X.; Yue, H.; Li, S.; Guo, J.; Guan, Z.; Zhu, Z.; Niu, B.; Zhang, T.; Wang, J. Genetic Polymorphisms in DNA Repair Gene APE1/Ref-1 and the Risk of Neural Tube Defects in a High-Risk Area of China. Reprod. Sci. 2021, 28, 2592–2601.
- Li, G.; Wang, X.; Wang, X.; Guan, Z.; Guo, J.; Wang, F.; Zhang, J.; Niu, B.; Zhang, T.; Wang, J.; et al. Polymorphism rs1052536 in Base Excision Repair Gene Is a Risk Factor in a High-Risk Area of Neural Tube Defects in China. Med. Sci. Monit. 2018, 24, 5015–5026.
- Larsen, D.H.; Stucki, M. Nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 2016, 44, 538–544.
- Shibata, A.; Jeggo, P.A. DNA double-strand break repair in a cellular context. Clin. Oncol. (R. Coll. Radiol.) 2014, 26, 243–249.
- Spies, J.; Polasek-Sedlackova, H.; Lukas, J.; Somyajit, K. Homologous Recombination as a Fundamental Genome Surveillance Mechanism during DNA Replication. Genes 2021, 12, 1960.
- Li, J.; Sun, H.; Huang, Y.; Wang, Y.; Liu, Y.; Chen, X. Pathways and assays for DNA double-strand break repair by homologous recombination. Acta Biochim. Biophys. Sin. 2019, 51, 879–889.
- Huselid, E.; Bunting, S.F. The Regulation of Homologous Recombination by Helicases. Genes 2020, 11, 498.
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Lobrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427.
- Lobrich, M.; Jeggo, P. A Process of Resection-Dependent Nonhomologous End Joining Involving the Goddess Artemis. Trends Biochem. Sci. 2017, 42, 690–701.
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282.
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506.
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211.
- Alt, F.W.; Schwer, B. DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair 2018, 71, 158–163.
- Wang, W.; Peng, M.; Yuan, H.; Liu, C.; Zhang, Y.; Fang, Y.; Su, Y.; Zhang, X.; Zhang, H.; Tang, Y.; et al. Studying the mechanism of sperm DNA damage caused by folate deficiency. J. Cell. Mol. Med. 2022, 26, 776–788.
- Zhao, J.; Guan, T.; Wang, J.; Xiang, Q.; Wang, M.; Wang, X.; Guan, Z.; Xie, Q.; Niu, B.; Zhang, T. Influence of the antifolate drug Methotrexate on the development of murine neural tube defects and genomic instability. J. Appl. Toxicol. 2013, 33, 915–923.
- Pei, P.; Cheng, X.; Yu, J.; Shen, J.; Li, X.; Wu, J.; Wang, S.; Zhang, T. Folate deficiency induced H2A ubiquitination to lead to downregulated expression of genes involved in neural tube defects. Epigenet. Chromatin 2019, 12, 69.
- Xie, Q.; Li, C.; Song, X.; Wu, L.; Jiang, Q.; Qiu, Z.; Cao, H.; Yu, K.; Wan, C.; Li, J.; et al. Folate deficiency facilitates recruitment of upstream binding factor to hot spots of DNA double-strand breaks of rRNA genes and promotes its transcription. Nucleic Acids Res. 2017, 45, 2472–2489.
- Wilson, M.P.; Hugge, C.; Bielinska, M.; Nicholas, P.; Majerus, P.W.; Wilson, D.B. Neural tube defects in mice with reduced levels of inositol 1,3,4-trisphosphate 5/6-kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 9831–9835.
- Hanakahi, L.A.; West, S.C. Specific interaction of IP6 with human Ku70/80, the DNA-binding subunit of DNA-PK. EMBO J. 2002, 21, 2038–2044.
- Hanakahi, L.A.; Bartlet-Jones, M.; Chappell, C.; Pappin, D.; West, S.C. Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair. Cell 2000, 102, 721–729.
- Herrera, E.; Samper, E.; Blasco, M.A. Telomere shortening in mTR−/− embryos is associated with failure to close the neural tube. EMBO J. 1999, 18, 1172–1181.
- Bekaert, S.; Derradji, H.; Baatout, S. Telomere biology in mammalian germ cells and during development. Dev. Biol. 2004, 274, 15–30.
- Tebbs, R.S.; Flannery, M.L.; Meneses, J.J.; Hartmann, A.; Tucker, J.D.; Thompson, L.H.; Cleaver, J.E.; Pedersen, R.A. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev. Biol. 1999, 208, 513–529.
- Xanthoudakis, S.; Smeyne, R.J.; Wallace, J.D.; Curran, T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8919–8923.
- Ma, Y.; Zhang, P.; Wang, F.; Yang, J.; Yang, Z.; Qin, H. The relationship between early embryo development and tumourigenesis. J. Cell Mol. Med. 2010, 14, 2697–2701.
- Pieroth, R.; Paver, S.; Day, S.; Lammersfeld, C. Folate and Its Impact on Cancer Risk. Curr. Nutr. Rep. 2018, 7, 70–84.
- Naghibalhossaini, F.; Mokarram, P.; Khalili, I.; Vasei, M.; Hosseini, S.V.; Ashktorab, H.; Rasti, M.; Abdollahi, K. MTHFR C677T and A1298C variant genotypes and the risk of microsatellite instability among Iranian colorectal cancer patients. Cancer Genet. Cytogenet. 2010, 197, 142–151.
- Paschalis, A.; Sheehan, B.; Riisnaes, R.; Rodrigues, D.N.; Gurel, B.; Bertan, C.; Ferreira, A.; Lambros, M.B.K.; Seed, G.; Yuan, W.; et al. Prostate-specific Membrane Antigen Heterogeneity and DNA Repair Defects in Prostate Cancer. Eur. Urol. 2019, 76, 469–478.
- Li, G.; Wu, J.; Li, L.; Jiang, P. p53 deficiency induces MTHFD2 transcription to promote cell proliferation and restrain DNA damage. Proc. Natl. Acad. Sci. USA 2021, 118, e2019822118.
- Schmidt, T.T.; Sharma, S.; Reyes, G.X.; Kolodziejczak, A.; Wagner, T.; Luke, B.; Hofer, A.; Chabes, A.; Hombauer, H. Inactivation of folylpolyglutamate synthetase Met7 results in genome instability driven by an increased dUTP/dTTP ratio. Nucleic Acids Res. 2020, 48, 264–277.