1. MMR and NTDs
The MMR pathway, including mutL homolog 1 (MLH1), MutS protein homologue 2 (MSH2), MutS homologue 6 (MSH6) and PMS1 homologue 2 (PMS2), is responsible for the surveillance and correction of errors during DNA replication, repair, and recombination. MSH2 couples with MSH6 or MSH3, forming MutSα or MutSβ complexes, respectively, and MLH1 couples with PMS2, PMS1 or MLH3, forming MutLα, MutLβ or MutLγ complexes, respectively [
70]. MutS and MutL are ultimately responsible for the recognition of mismatches and insertion–deletion loops, after which the MLH1/PMS2 complex is recruited to degrade the mutated stretch and initiate resynthesis [
71,
72] (
Figure 4).
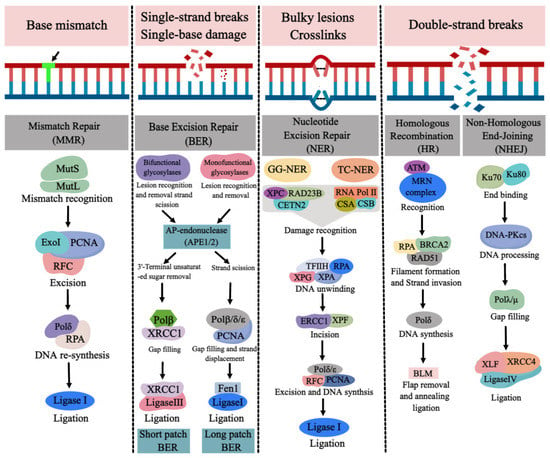
Figure 4. The major types of DNA repair pathways. Base mismatch is repaired by mismatch repair (MMR), single-strand breaks (SSBs) and single-base damage is repaired by base excision repair (BER). Bulky lesions and crosslinks are repaired by nucleotide excision repair (NER) and double-strand breaks (DSBs) are repaired by homologous recombination (HR) and nonhomologous end joining (NHEJ). (Created with
https://image.medpeer.cn, accessed on 30 November 2022).
Microsatellite instability (MSI) can reflect the instability of the genome, which is caused by inactivating a series of DNA MMR genes, especially
MLH1 [
73]. Moreover, MSI is related to hypermethylation of the
MLH1 promoter region (which can lead to gene inactivation) in tumors; thus, MSI can reflect the deletion or dysfunction of the MMR system [
74]. Studies have shown that folate has a role in regulation of the MSI. A study of folate levels and MSI statuses in the colonic mucosa of 26 patients with chronic ulcerative colitis found that MSI was found in 3/23 patients (13%) with ulcerative colitis. The three patients with MSI exhibited lower levels (30–50%) of folate concentrations in their serum, whole blood and colonic mucosa. Interestingly, after one of the patients with MSI received a folate supplement (5 mg/day) for 6 months, three of six microsatellite markers became stable, suggesting a close relationship between folate status and MSI [
75]. Liu et al. studied the genomic instability in human fetuses with NTDs under folate deficiency; the results demonstrated that the total positive rate of MSI in the NTDs group was 46% (23 of 50 NTD patients) and
hMLH1 promoter demonstrated two specific methylation patterns at “4,5 CpG” sites in the target region, which were a 180-bp fragment upstream from exon-1 of
hMLH1 (−271 to −92 bp, containing 11 CpG sites) in some NTD samples [
76]. These results suggest that folate-deficiency-induced occurrence of NTDs were associated with the abnormal function of the MMR system. It has been reported that H3K36me3 (histone H3 trimethylated lysine 36) recruits MutSα (MSH2-MSH6) onto chromatin through direct interactions with the MSH6 PWWP domain to regulate the MMR in vitro [
77].
Hypb encodes the H3K36me3 specific methytransferase, which is closely related to NTDs, as observed by knocking it out in mouse embryos [
78]. The targeted sequencing analysis of human NTDs with folate insufficiency found that a great majority of variants found were single-nucleotide substitutions or base–base mismatches that were repaired by MMR, and the most potentially deleterious variants were observed within H3K36me3 occupancy regions (68.1% of total occurrences). The co-immunoprecipitation and MSI analysis found that low folate status attenuated the binding of Msh6 with H3K36me3 and resulted in impairment of the MMR pathway, suggesting that folate-deficiency-induced NTDs in humans could be mediated via an adverse impact on MMR [
79]. The methy-CpG binding domain protein 4 (MBD4) recognized T/U or T/G mismatch in the methylated CpG nucleotide sequence, which appeared to coordinate MMR with DNA methylation events. The C-terminal domain of MBD4 interacted with MLH1, which was involved in MMR [
80,
81,
82]. It has been reported that
Mbd4 gene and
Ift122 gene share the same starting sequence (Exon 1) at the murine locus, and
Mbd4 exon 1–3 mutation abrogates the expression of both
Mbd4 and
Ift122. Mouse embryos defective in
Ift22 gene (Exon 1–3) ranging from E10.5 to E13.5 demonstrated a complex phenotype characterized primarily by failure of neural tube closure (such as exencephaly) [
10]. It was speculated that the
Mbd4 gene might interact with the
Ift122 gene at the transcriptional level and participate in the formation of mouse embryonic malformation. Therefore, the alteration of epigenetic modification (methylation of DNA or histone) induced by folate deficiency or mutation in MMR genes contributes to the closure of the neural tube.
2. BER and NTDs
The BER pathway is a highly conserved process to repair the oxidative damage, alkylation, deamination and methylation of DNA bases in cells [
83,
84]. DNA glycosylases in BER pathway recognize and remove base damage in three types: monofunctional, bifunctional, and nei-like DNA glycosylases [
85]. Monofunctional enzymes such as uracil DNA glycosylase (UNG), single-strand selective monofunctional uracil DNA glycosylase (SMUG1), G/T mismatch-specific thymine DNA glycosylase (TDG), MBD4, N-methylpurine DNA glycosylase (MPG), and adenine DNA glycosylase (MUTYH) excise the damaged base, leaving an apurinic/apyrimidinic (AP) site and a phosphodiester backbone, which is further hydrolyzed by AP endonuclease1 (APE1), forming a single-strand break (SSB) with 5′-deoxyribosephosphate and 3′-hydroxyl ends [
85,
86]. Bifunctional glycosylases, including endonuclease-like protein 1 (NTH1) and 8-oxoguanine DNA glycosylase 1 (OGG1), remove the base and cleave the phosphodiester bond, leaving a β-elimination, which is hydrolyzed by APE1 and creates a nucleotide gap. Nei-like DNA glycosylases (NEIL1, NEIL2, NEIL3) catalyze a β/δ-elimination reaction, where the phosphodiester bond is cleaved, and a 3′-phosphate group is generated by polynucleotide kinase phosphatase (PNKP) [
87,
88]. The poly (ADP-ribose) polymerase 1(PARP1), as a DNA damage sensor, protects the strand break and recruits the DNA repair protein by its poly (ADP-ribosyl)ation activity [
89]. DNA polymerase β (Polβ) in BER pathway fills the gap and removes the 5′-deoxyribosephosphate [
90]. Finally, DNA ligase IIIα (LigIIIα) interacts with X-ray repair cross-complementing protein 1 (XRCC1) to complete the short-patch BER, which is responsible for repairing the single nucleotide gap [
91,
92]. Long-patch BER is employed to repair the gap of 2–10 nucleotides by proliferating cell nuclear antigen (PCNA)-dependent DNA polymerases δ/ε (Polδ/ε) to displace the damaged strand. Flap endonuclease 1 (FEN1) removes the displaced strand, leaving a nick ligated by the DNA ligase I (LigI) [
93,
94] (
Figure 4).
The BER pathway is necessary for normal brain development and is closely related to neurodevelopmental disorder [
95]. Folate deficiency results in DNA damage accumulation due to incapacitation of the BER [
96,
97,
98]. The effect of folate deficiency on BER shows that the balance and coordination of BER in mice are impaired by increasing the UDG protein level (30%) without subsequently stimulating the APE1 and Polβ, resulting in a greater accumulation of DNA damage in response to folate deficiency [
96]. Folate deficiency impacts BER capacity by inhibiting Polβ expression at the transcription level through the negative regulatory factors binding to the folate response region within the core promoter of Pol β in mice [
97]. These studies indicate that folate deficiency decreases the capacity of BER by affecting the expression of the key enzymes in BER, which might contribute to the occurrence of NTDs. Olshan et al. conducted a case–control analysis from 250 NTDs (including 125 spina bifida, 125 oral clefts) and 350 non-malformation controls to investigate the relationship between DNA repair gene polymorphisms and NTDs; the results show that the polymorphism of BER gene
APE1 (Asp148Glu) reduced the risk of spina bifida, while, on the other hand, the BER genes
XRCC1 (Arg399Gln) and
OGG1 (Ser326Cys) could increase the risk of spina bifida [
10]. Our previous study found that the polymorphisms (rs3136817, rs77794916, and rs1760944) of BER gene
APE1 in fetal brain tissues from 165 NTD fetuses and 300 control fetuses were statistically associated with NTDs under low folate status for Han population in a high-risk area of China. The allele C of rs3136817, allele T of rs77794916, and allele G of rs1760944 were associated with an increased risk for encephalocele (
OR = 2.52, 95%
CI, 1.25–5.07;
OR = 1.80, 95%
CI, 1.04–3.12; and
OR = 1.96, 95%
CI, 1.12–3.45), compared with those harboring the alleles T, C, and T, respectively [
99]. Meanwhile, a case–control study on the polymorphisms of BER gene
LIG3 in 108 NTD pregnant women and 233 normal healthy pregnant women found that the TT genotype of rs1052536 in
LIG3 increased the risk of anencephaly (
OR = 2.69, 95%
CI, 1.18–6.10) and T allele carriers had significantly increased risk of cranial NTDs (
OR = 1.56, 95%
CI, 1.04–2.35) [
100]. These results indicate that BER gene mutation might be a potential genetic risk factor for NTD occurrence.
3. DSBR and NTDs
DNA double-strand breaks (DSBs) are the most serious type among all forms of DNA damage and are repaired by homologous recombination (HR) and nonhomologous DNA end joining (NHEJ) [
101,
102]. HR begins with the binding of the MRN complex (MRE11, RAD50 and NBS1) to the broken DNA ends [
103]. MRN interacts with CtIP, promoting end resection [
104]. Bulk resection occurs via activation of the nucleases exonuclease 1 (EXO1), DNA replication ATP-dependent helicase (DNA2), and the Bloom syndrome helicase (BLM), generating single-stranded DNA (ssDNA), which is rapidly bound by RPA. The RAD51-ssDNA nucleofilament is formed by replacing RPA with RAD51 [
104]. The RAD51-ssDNA filament carries out homology search, then invades a homologous DNA sequence and forms a displacement loop (D-loop) [
104]. DNA polymerase can subsequently add nucleotides at the free 3′end to restore the sequence of DNA damage [
105]. In NHEJ, the DNA ends in DSBs are recognized by the Ku70/Ku80 heterodimer, which then recruits and activates the catalytic subunit of the DNA dependent protein kinase (DNA-PK). The compatible ends are created by nucleases such as Artemis [
106,
107]. The DNA ligase IV and XRCC4 complex ligate the ends [
108,
109,
110] (
Figure 4).
The DSBR pathway plays potential roles in neurodevelopment and neuronal function [
111]. DSBR is also influenced by folate status. Folate deficiency increases the expression of (phosphorylated H2AX) γ-H2AX, a DNA injury marker protein, as well as the methylation frequency of CpG site in the DSBR gene
Rad54 promoter region, and downregulates the expression of Rad54 in mouse sperm and spermatocyte line with folate deficiency [
112]. The study of pathogenesis of NTDs has demonstrated that the E3 ligase mouse double minute 2 homolog (Mdm2) colocalized with γ-H2AX is recruited to the sites of DSBs in mouse embryonic stem cells (mESCs) after exposure to methotrexate (a folate antagonist [
16]), which might contribute to NTDs induced by folate deficiency [
113]. The DSBs in mESCs with folate deficiency were most prominently enriched in intergenic regions of the genome (45.7%), followed by the intronic and exonic regions (25.2% and 23.24%, respectively) [
15]. These studies indicate that DSBs caused by folate deficiency is predisposition for NTDs. Furthermore, it was reported that the DSBR gene
XRCC3 (Thr241Met) polymorphism reduced the risk of spina bifida from a case–control study by the California birth defects monitoring program [
10]. Inositol-5,6-kinase 1,3,4-triphosphate (ITPK1) is a key enzyme for inositol hexaphosphate (IP6) synthesis. Down-regulating the expression of
ITPK1 in transgenic mice through gene capture technology induced NTDs, bone defects, and growth retardation [
114]. IP6 can activate NHEJ by binding to Ku protein in a DNA-PK complex and changing its conformation [
115,
116]. Therefore, the occurrence of NTDs caused by the down-regulation of ITPK1 might be caused by the reduction of IP6 synthesis, which eventually leads to the obstruction of the NHEJ process. Telomerase RNA knockout (
mTR−/−) mice exhibited NTDs, which is related to telomere deletion, chromosomal instability, and apoptosis [
117]. In
mTR−/− mice, cells lacking Ku70/86 protein were more prone to apoptosis. Ku70/86, a member of the DNA-PK complex, is involved in NHEJ, which can repair dysfunctional telomeres [
118]. It is speculated that loss of Ku protein will cause NHEJ disorder, resulting in abnormal apoptosis and eventually leading to NTDs. These studies suggest that both DSB induced by folate deficiency and DSBR gene mutation are related to NTDs.
Although DNA repair pathways (such as BER, MMR, DSBR) have been studied for many years, a complete understanding of how these pathways function in the pathogenesis of NTDs is still lacking. Given the intricate complexity of human NTDs, delineation of the relationship between folate and DNA repair pathways during neural tube closure remains an attractive field. Understanding how impairment of the regulatory mechanisms of DNA repair pathways may increase the risk of NTDs will help develop novel therapeutic approaches to reduce NTD occurrence in future. Here, the gene mutations in DNA repair pathways most likely lead to NTDs during the period of neural tube closure. Other malformations were also observed at the early formation of critical organ systems. The BER gene
Xrcc1 mutant embryos demonstrated increased cell death in the epiblast and an altered morphology in the visceral embryonic endoderm at embryonic day 6.5–7.5 [
119]. The degeneration in
Apex1 gene homozygous mutant embryos were observed at embryonic day 5.5 [
120]. These data suggest that mutations in DNA repair genes leads to dysfunction in certain tissues or organs and disturbed genomic integrity during the embryonic period, resulting in embryo developmental anomalies. Folate deficiency might cause mutations or abnormal methylation of the gene in DNA repair pathways, promote genomic instability, and alter gene expression. This might impair the balance of apoptosis and proliferation during the development of the neural tube. DNA repair pathways might be a link between folate deficiency and NTDs.
In recent years, there have been few studies focusing on the mechanism of the DNA repair pathway and NTDs with folate deficiency. Studies have shown that folate insufficiency promotes increase of rare variants through the loss of MMR, resulting in more severe MMR deficiency, providing a mechanistic link between low folate status and the occurrence of NTDs through deficits in MMR machinery [
79]. It has been reported that biological behavior is similar between tumorigenesis and early embryonic development [
121]. Some studies found that alteration of folate-metabolism-related enzymes affecting genomic stability was closely related to the function of DNA repair pathways in cancers, such as methylenetetrahydrofolate reductase (MTHFR) [
122,
123], prostate-specific membrane antigen (PSMA) [
124], mitochondrial methylenetetrahydrofolate dehydrogenase (MTHFD2) [
125] and Folylpolyglutamate synthetase (FPGS) [
126]. Therefore, mutation in DNA repair pathways or folate-metabolism-related enzymes could cause genomic instability, impair the balance of cell proliferation and apoptosis, and, finally, lead to the occurrence of NTDs.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24032220