Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Hematology
Tyrosine kinase inhibitors (TKIs) have been extensively used as a treatment for chronic myeloid leukemia (CML). Dasatinib is a broad-spectrum TKI with off-target effects that give it an immunomodulatory capacity resulting in increased innate immune responses against cancerous cells and viral infected cells.
- dasatinib
- tyrosine kinase inhibitors
- CML
- cancer
1. Introduction
1.1. Dasatinib
Dasatinib is a broad-spectrum tyrosine kinase inhibitor originally developed to treat chronic myeloid leukemia (CML) [1]. CML is a hematopoietic progenitor cell leukemia, in which overgrown myeloid cells accumulate in bone marrow and peripheral blood [2]. A translocation between chromosomes 9 and 22 results in an aberrant chromosome 22 (or Philadelphia (Ph) chromosome), generating the BCR-ABL oncogene. BCR-ABL constitutively activates tyrosine kinases (TK) that drives both Ph+ CML and Ph+ acute lymphoblastic leukemia (ALL) [3]. Treatment for CML was drastically improved with imatinib, one of the first TK Inhibitors (TKI) targeting BCR-ABL TK [4]. Furthermore, development of these small molecules led to the development of alternative inhibitors such as the second-generation drug dasatinib, which yield up to 20-300 times higher activity and has faster and deep molecular response (DMR) than imatinib [1].
Dasatinib has other off-target effects inhibiting other TK (Table 1) [5] with potential side effects such as hematological, pulmonary and gastrointestinal toxicity that could limit its clinical use [6]. Interestingly, off-target effects have been related to potential benefits such as an increased natural killer cell (NK)-mediated cytotoxic capacity against cancerous cells and viral infected cells [7] and an anti-aging capacity. The potential immunomodulatory effects of dasatinib on NK cells and other innate cells and its therapeutic role against CML, HIV infection and aging (Figure 1).
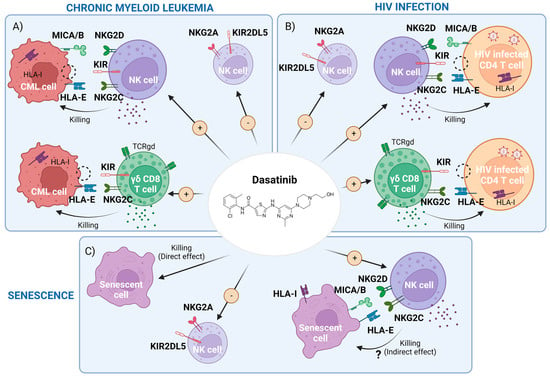
Figure 1. Scheme of dasatinib-mediated effects that may interfere with chronic myeloid leukemia (CML), HIV-1 infection and cellular senescence. (A) CML cell killing by the enhancement of memory natural killer (NK) cells and cytotoxic CD8+ T cells expressing γδTCR and a reduction of inhibitory receptors driven by dasatinib. (B) HIV infected cells’ clearance, carrying pro-viral DNA, by dasatinib activity through the potentiation of the above cell subpopulations. (C) Representation of both direct killing and a potential indirect effect by boosting NK and CD8+ cells of senescent cells with dasatinib. Figure made with BioRender.com (accessed on 7 March 2023).
Table 1. Tyrosine kinases (TKs) targeted by dasatinib and the cellular processes in which they are involved. Adapted from Araujo et al., 2010 [5]. Abbreviations: PDGFRβ, platelet-derived growth factor receptor beta; EPHA2, ephrin type-A receptor 2; EFNB, ephrin-B; M-CSF, macrophage colony-stimulating factor.
| Tyrosine Kinase Target (s) | Pathways and Processes |
|---|---|
| SRC family (SRC, LCK, YES, FYN) | Oncogenic, invasive and bone-metastatic processes |
| BCR-ABL | Promotion of growth advantage of leukemic cells |
| c-KIT | Cell growth |
| PDGFRβ | Tumor growth capacity and cell survival |
| c-FMS | Macrophage behavior regulation by M-CSF |
| EPHA2 receptor | Interference with EFNB-dependent suppression of apoptosis/Cell behavior |
1.2. Natural Killer Cell Biology
Natural killer cells (NK cells), are cells from the innate immune system that show strong cytolytic function against stressed cells such as tumoral cells and virus-infected cells. NK activation state is determined by a balance of multiple activation and inhibition signals mediated by NK inhibitory and activating receptors that bind to NK ligands from other neighboring cells (Figure 2A). Most of these ligands are HLA class I molecules, such as HLA-A, B, C and HLA-E. Depending on the balance between these NK receptors (NKR) and their HLA ligands, NK cells will either be activated to kill the target cell or inhibited, allowing the target cell to survive. NK cells are an heterogeneous population harboring multiple subsets that differentially expressed NK receptors such as: killer cell immunoglobulin-like receptors (KIRs), natural killer group 2 such as NKG2A, NKG2C and NKG2D, and natural cytotoxicity receptors (NCRs), for example NKp30, NKp40 or NKp46 [8,9]. The main activating and inhibiting NK receptors and their respective target cell ligands are shown in Figure 2A. The NKG2C and NKG2D receptors are activating NK receptors that could detect abnormal or malignant cells by binding with HLA-E and the MIC A/B ligands, respectively. These interactions potentiate NK cell killing [10,11] (Figure 2A).
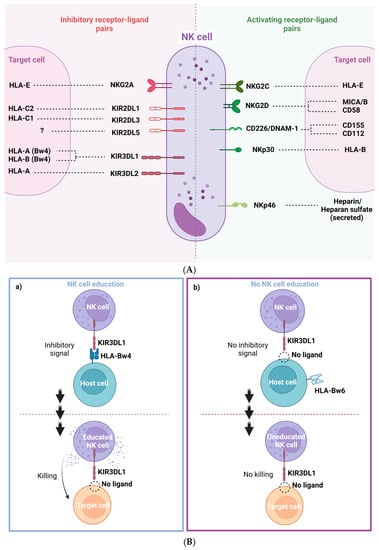
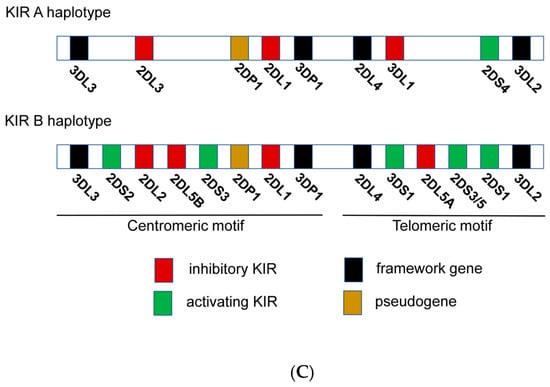
Figure 2. (A) Representation of NK cell inhibitory and activating receptor-ligand pairs. (B) NK cell education process and “missing self” killing of target cells. (a) NK cells are recognized through KIR3DL1 receptor host cells expressing HLA-Bw4 and become educated to recognize its absence then in target cells. This process leads to an efficient target cell killing. (b) Absence of recognition of HLA-Bw4 by KIR3DL1 drives a deficient cell killing of target cells with “missing self” by miseducation of the NK cell. (C) Genomic organization of KIR A and B haplotypes.
NK cells are activated by target cells that down regulate “self” HLA. This NK activation mechanism appears by a phenomenon known as education or licensing. Education requires the interaction between inhibitory NK cell receptors and their own HLA ligands [13,14]. Educated NK cells remain inactive by the interaction of NKRs inhibitor with autologous HLA-expressing neighboring cells. However, NK cells can also be activated by viral infected cells and abnormal or injured cells that usually have a lower HLA class I expression to evade cytotoxicity driven by conventional CD8+ T cells. NK cells can detect these anomalous target cells by the lack of inhibiting interaction (low o non-inhibitory HLA interactions). This phenomenon is called “missing self” detection and generally results in NK cell activation, degranulation and target cell death (Figure 2B). For NK cell activation, additional activating signal are necessary [15]. NK cells are activated when the ratio of inhibitory and activator NKR signaling favors activation [9] (Figure 2B). Many inhibitory KIRs from NK cells interact with HLA alleles. KIR and HLA proteins are both encoded by highly polymorphic alleles, enabling a wide diversity of receptor/ligand interactions. Education through inhibitory receptors sensitizes NK cells to detect “missing self” cells, while education through activating receptors inhibited NK cell cytotoxicity [16] (Figure 2B).
Despite the high level of KIR gene content variability, there are two main groups of KIR haplotypes, termed “A” and “B” (Figure 2C). The A haplotypes include inhibitory KIR genes coding for receptors such as KIR3DL1, which recognize HLA-A and HLA-B molecules that contain Bw4 epitopes. The B haplotypes include several activating KIR genes such as KIR3DS1 (Figure 2C). Then, the KIR genetic combination can be AA, AB or BB genotype. There are two possible established epitopes for HLA-B: Bw4 or Bw6. KIR3DL1 is highly polymorphic and, depending on the haplotype, NK cells can express: high, low, or null levels of KIR3DL1 [17]. In fact, KIR3DL1 and HLA-B genetic polymorphisms regulate NK cell function. Their stronger ligand affinity correlates with enhanced NK cell inhibition but also with increased education and, consequently, potentiates “missing self” NK capacity to detect and kill anomalous cells [18] (Figure 2B).
The inhibitory NKG2A receptor binds to HLA-E molecules. Signal peptides from the leader sequence of HLA-A, -B and -C proteins, which are codified by the first exon of the MHC gene, must bind to HLA-E in order to fold properly and reach the cell membrane and to become a ligand for the inhibitory NKG2A receptor. In addition, the anchor residue of the nonamer peptides that bind to HLA-E corresponds to residue -21 of the classical HLA class I leader sequence. There are two amino acid variations at position -21, a methionine (-21M) or a threonine (-21T) [19,20]. HLA-B antigens can have both -21M and -21T variants, depending on the HLA-B haplotypes that are associated with two NK cell education profiles. On the one hand, HLA-B -21M alleles contribute more effectively to NKG2A-mediated education, which suppresses NK cell activation when the target cell expresses stabilized levels of HLA-E. HLA-B -21T alleles mainly contribute to education through inhibitory KIRs, especially KIR3DL1 [15], as HLA-B -21T and HLA-Bw4 are genetically linked. Then, the HLA-Bw4 alleles are, in fact, HLA antigens encoded by -21T, able to interact and educate through KIR3DL1/HLA-Bw4, allowing the NK cell cytotoxicity upon “missing self” recognition [9,20] (Figure 2B).
2. Dasatinib-Mediated Immunomodulatory Effects in CML
2.1. Dasatinib Effect on NK Cells and Innate T Cells
Besides its direct impact on leukemic cells through inhibition of constitutive tyrosine kinase activity, dasatinib’s off-target effects include: inhibition of proliferation and activation of T cells, and in vitro suppression of cytotoxic activity of NK cells [21,22] This is due to the potent inhibition of several off-target kinases of the Src family, such as Lck and Fyn in T cells [23]. TKI treatments can restore an anti-leukemic effector function, such as specific cytotoxic T lymphocytes’ (CTL) responses against leukemia-associated antigen (LAA). Overall, this results in a reduction in leukemic cell load major molecular remission (MMR) and a better recovery [24,25]. Dasatinib has the potential to reduce Tregs and derived factors (sCTLA-4), especially in patients developing large granular lymphocytes (LGLs), lymphocytosis [26] and NK cell differentiation, promoting immune stimulation [27]. This is relevant because the proportion of Treg cells is abnormally elevated in CML individuals at diagnosis, compared to healthy controls. Furthermore, dasatinib also reduces myeloid-derived suppressor cells (MDSC) in CML patients [28].
Up to half of the patients receiving dasatinib treatment develop an LGL lymphocytosis, mainly composed of cytotoxic cells (NK cells and CD8+ γδ T cells) [29,30] but this also includes CD57+ cytotoxic CD4+ T cells with anti-leukemic properties [31]. This LGL expansion is associated with an improved anti-leukemic response in both CML and Ph+ ALL patients [29,32,33,34,35]. The CD8+ T cell response in dasatinib-mediated LGL response includes TCR-Vβ+ expansion of either oligoclonal or polyclonal origin with cells resembling healthy memory CD8+ T cells [36]. Importantly, dasatinib may have an immunomodulatory role in non-conventional or innate T cells by increasing cell number, activation status and Th-1 polarization of innate αβ iNKT-cells in treated CML patients [37]. In addition, it has been reported that dasatinib increased a novel innate subset termed innate CD8+ T-cell-expressing IFNγ [37].
CML patients treated with dasatinib presented more classical NK cells (CD3−CD56+) and matured NK cells (CD56+CD57+) compared to imatinib- or nilotinib-treated patients [38]. These patients also presented lower expression of NK-inhibitory markers (KIR2DL5A, KIR2DL5B and KIR2DL5), which was associated with an MMR [39] and an increase in KIR2DL1 expression [8].
Dasatinib promoted NK-cell cytokine expression and cytotoxic activity towards the CML-derived cell line K562 [18,40], especially when KIR3DL1/HLA-Bw4 interactions were null, low or weak. Of note, Izumi et al. showed that blocking KIR3DL1/HLA-Bw4 binding with an anti-KIL3DL1 antibody potentiated NK cytotoxicity [18]. Furthermore, Shen et al. reported that this increased NK cytotoxic activity can be emulated in vitro using dasatinib treatment after IL-2/IL-15-mediated expansion of NK cells. In this setting, dasatinib also increases the percentage of NKG2A−CD57+ NK cells and the expression of activating receptors CD226 (DNAM-1), NKp46 and NKG2D [41], promoting their capacity to kill by degranulation of the CML cell line K562, not expressing HLA class I. Finally, dasatinib-mediated reduction of NKG2A also may boost NK cytotoxicity and improve MMR [18,42].
2.2. Dasatinib Increases Memory-like Natural Killer (NK) Subsets Displaying Activity against Both Leukemic and Cytomegalovirus (CMV) Infected Cells
There is discussion on whether development of LGLs with dasatinib is associated with previous immunity to CMV [32,43] or related to CMV viral load (VL) [7,43]. Ishiyama et al. reported that NK cells are the main component of LGLs in patients with CML treated with dasatinib, and NK cell expansion was highly associated with CMV-serostatus [7]. The authors performed a principal component analysis (PCA) with multiple markers on NK cells after dasatinib treatment and determined that NK cells from CMV+ individuals had a CMV-associate phenotype, named memory-like NK cells, with highly differentiated NK phenotypes (NKG2ChighNKG2AlowCD57highLIR-1highNKp30lowNKp46low). NK cells from CMV-uninfected individuals were negative for this CMV-related signature [7,44]. Remarkably, a higher grade of NK cell differentiation at CML diagnosis predicts both a greater expansion of CMV-related memory-like NK cells and a lower leukemic-cell load after dasatinib treatment in CMV+CML patients [7,44]. Kadowaki et al. reported that a persistent, low-level CMV replication, often subclinical, triggered memory-like NK cell expansion. This suggests that CMV may trigger NK-cell expansion and both leukemia and dasatinib are enhancing factors that expand this NK subpopulation [45]. This hypothesis was reviewed by Climent et al. [46]. Importantly, this NKG2C+CD57+ subset of memory-like NK cells may represent NK cells with unique adaptive and editing properties [44]. Thus, the expansion of this subset could promote long-term memory and high cytotoxic activity against CML, even after dasatinib treatment interruption [45]. Recent studies have also observed an expansion of CD56neg NK cell populations exclusively in CMV+ patients treated with dasatinib, and this increase parallels memory NKG2C+CD57+ NK cells [47]. These authors propose that CD56− NK cells may be an exhausted population induced by chronic activation through CMV reactivation but, paradoxically, are proposed as a hallmark of CML control because it predicts a better clinical outcome. Overall, these results suggest that dasatinib immunomodulatory effects on NK cell responses against CMV could also be relevant against malignancies or other viral infections [48], such as HIV infection [46].
2.3. CML Control and Therapeutic Treatment Interruption: Immunological Factors Involved in a Successful Treatment-Free Remission (TFR)
Once a DMR is observed, TKI treatment can be interrupted in selected patients with the aim of achieving a TFR [49]. Dasatinib and nilotinib treatments are associated with a stronger DMR, increasing the chances of longer TFR [49]. A better understanding of the factors that could predict longer TFR is of paramount importance [50,51,52]. Moreover, half of the individuals who stopped treatment were able to keep TFR during at least one year and developed high levels of NK cells [53,54,55] and neutrophils [56] but not T cells [53,57] indicating that these populations are key to keeping CML under control.
Regarding the implication of NK cells in TFR, individuals who controlled CML after imatinib therapy cessation showed higher NK cytotoxic function towards the target K562 cell line, lacking HLA class I. Furthermore, NKG2D gene polymorphisms [58] and the IFN-γ and TNF-α cytokine secretion by NK (CD56dimCD16−) cells correlated with the successful drug discontinuation and control of CML [57,59]. Increased mature (CD57+) and cytotoxic (CD16+ and CD57+) NK cells, together with IFN therapy prior to TKI cessation, have been also shown to produce better CML outcomes after treatment interruption [60]. IFNα treatment also increased differentiated NKG2C+ NK cells, increased NKp46 expression on the CD56bright/CD16- NK cell subpopulation and modulated NK cell cytotoxicity [61].
TFR has also been evaluated in dasatinib stopping TKI trials [62]. The DADI study [53,63] demonstrated that high levels of NK cells (CD56+), LGL NK cells (CD56+CD57+) and low levels of Treg (CD25+CD127low) preceding the dasatinib interruption were associated with longer TFR periods. Furthermore, a critical role of Treg inhibition by dasatinib, potentiating NK cell function, promotes a DMR [64]. Likely expanded NK cell functionality and lower Treg frequencies may decrease the probability of a worse outcome after dasatinib interruption and result in longer TFR [65].
Recent studies suggest that the features most consistently linked to longer TFR, independently of the type of TKI treatment stopped, are: (1) a high frequency of cytotoxic subsets such as NK, NKT and CD8+ γδ T cells [66,67]; (2) high level of NK-activating receptors such as NKG2D, NKp30 or NKG2C on NK [11,67,68] and NKT cells [67]; (3) enhanced expression of activation cytokines or granzyme B in NK cells after stimulating with HSP70; and (4) KIR homozygosis at haplotype AA, which includes KIR3DL1. These hallmarks may be useful as prognostic biomarkers of longer TFR [67]. Further clinical trials are needed to test these predicting biomarkers for TFR [69]. Moreover, the memory-like NK cells, characterized as CD3−CD56dimCD57+ NKG2A− NKG2C+, were increased in patients with TFR success [70].
Results concerning NKG2A expression on NK cells are controversial [11]. Several studies suggested that low levels of NKG2A expression in NK cells is associated with longer TFR [70] and better CML prognostic [42]. In stark contrast, Xu et al. reported that an elevated expression of NKG2A in NK cells, especially in the CD56bright subset, was a good prognostic biomarker for TFR [11], as also reported by Vigón et al. [67]. The inhibitor receptor NKG2A has a dual function on NK cells: firstly, it has a key function in the NK education process; second, after inhibition of NK cell activation it could send other signals to NK cells [71]. In fact, we recently found that high expression of NKG2A is present in NK cells able to kill cancerous cells such as reprogrammed cells or those that downregulate HLA-E. Reprogrammed cells express Yamanaka factors and this gives them the capacity of being pluripotent embryonic stem cells (e.g., teratoma-like cells). These educated NK cells are able to kill by the missing self-recognition [72]. NKG2A expression could not be per se detrimental to the function of all NK cell subsets. Altogether, the increased expression of NKG2A in the CD56bright subset could be interpreted as an increased killing capacity against target cells with lower expression of HLA-E, such as cancerous cells or CML cells [11].
2.4. NK Immunogenetics Associated with CML Control or TFR
The diversity of allotypes of KIR3DL1 and HLA-Bw4 is associated with the receptor/ligand avidity and the NK cytotoxic capacity [12,17,73,74]. KIR genotypes have high genomic variations or allotypes that are associated with NK cell cytotoxic activity against CML and extended TFR periods [75,76]. Some reports suggested that CML patients with KIR2DL5B and KIR2DL2 alleles reached higher DMR after TKIs, implying that some KIR alleles or a specific combination of KIR genes can modify NK cell activity against CML cells [77,78]. In fact, KIR AA haplotypes, which include many KIR inhibitors such as KIR3DL1, are associated with better outcomes in TKI-treated CML patients [77] and are also linked to patients with sustained TFR [8,75]. It is interesting that the A haplotype including KIR3DL1 is highly associated with NK education (Figure 2B,C). Interaction of KIR3DL1 and HLA-Bw4 could affect NK cell education and cytotoxicity. In fact, the KIR3DL1*005 allele was highly linked with DMR, suggesting the relevance of this specific KIR3DL1. Consistently, DMR was coupled with higher NK cell killing in vitro in a NK cell cytotoxic assay against the CML cell line K562 without HLA class I, indicating that these educated NK cells could contribute to eliminating CML cells in vivo (Figure 2B). However, verification is needed, and the specific A haplotype KIR gene with the greatest impact needs to be identified [12] (Figure 2B). It has been shown that haplotypes of KIRs and HLAs were linked to a better outcome in a Japanese cohort [18,76]; specifically, Ureshino et al. reported that TFR in patients with HLA-Bw4 was higher than in patients with HLA-Bw6 alleles [79]. Similarly, HLA-Bw4 has been related to HIV control [80]. Altogether these genetic associations suggest that NK cells from patients achieving DMR or TFR could be better educated by interaction between the inhibitor receptor KIR3DL1 and HLA-Bw4, allowing activation of NK cells against cancerous cells that downregulate HLA expression, triggering “missing-self activation” (Figure 2B). Clinical trials with large cohorts are needed in order to explore deeply the NK immunogenetic factors associated with CML control [12].
2.5. Dasatinib as an Immunomodulator in Other Therapeutic Strategies against Cancer
The immunomodulatory activity of dasatinib has also been evaluated in combination with other therapeutic strategies against advanced malignancies such (1) immunotherapy with immune checkpoint inhibitors, where dasatinib immunomodulatory capacity has been investigated in increased programmed cell death protein 1 (PD-1) and programmed cell death ligand 1 (PD-L1) (PD1-PDL1) immunotherapy [81,82]; or (2) with chimeric antigen receptor (CAR)-engineered T cells (CAR-T). In CAR-T therapies, T cells are ex vivo modified by adding a gene for a receptor that helps the T cells to target specific myeloid antigens. A combined CAR-T and TKI approach has also been evaluated in some studies to enhance antitumor immunity and demonstrated that dasatinib limits CAR-T cells’ therapy side effects, such as the cytokine release syndrome (CRS) [83], and increases the anti-leukemia activity of CAR-T cells by decreasing cell exhaustion [84].
2.6. Summary of the CML Section
In brief, these findings support the idea that dasatinib contributes to better treatment response in CML patients through enhancement of the immune system, particularly via NK cell differentiation. CML patients with better outcomes could have done better due to genetic factors, such as AA alleles (homozygosis at KIR3DL1) and HLA-Bw4 associated with educated and highly cytotoxic NK cells able to detect malignant cells. Consequently, dasatinib could be useful, especially in the patients that do not have these protective features, to enhance cytotoxic activity of NK cells against CML cells by increasing memory-like NKG2C+CD57+ NK cells [70], γδ T cells and other innate CD8+ T cells [7,30,37,44]. In addition, these innate cells could express high levels of the activation receptor NKG2C, NKG2D, NKp46 or DNAM-1 and downregulate some inhibitory receptors such as NKG2A and KIR2DL5 [7,39,41,44] (Figure 1A).
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics15030917
This entry is offline, you can click here to edit this entry!