Muscle fatigue is defined as a decrease in maximal force or power generated in response to contractile activity, and it is a risk factor for the development of musculoskeletal injuries. One of the many stressors imposed on skeletal muscle through exercise is the increased production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which intensifies as a function of exercise intensity and duration. The progressive reduction in muscle fibers’ ability to generate force originates at different levels of the motor system and can be categorized into two types, namely, central and peripheral fatigue. Peripheral mechanisms of fatigue refer to activity-induced mechanical failure through processes at or distal to neuromuscular junctions, so they can be attributed to neuromuscular transmission and excitation–contraction coupling.
- muscle fatigue
- exercise
- oxidative stress
- antioxidants
1. Peripheral Muscle Fatigue
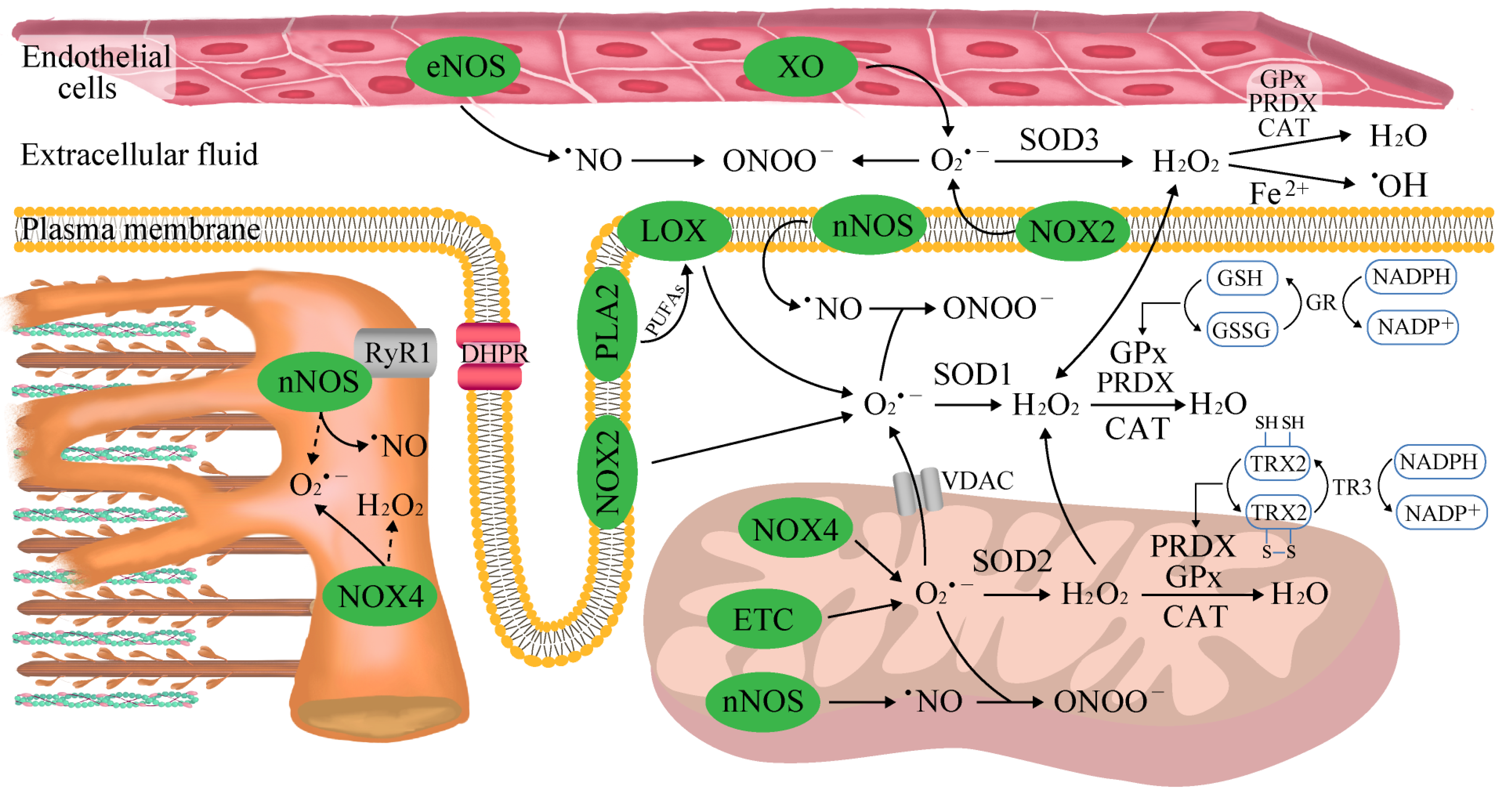
2. The Sources of Reactive Oxygen and Nitrogen Species (ROS/RNS) in Skeletal Muscle
3. Aerobic Exercise
4. Anaerobic Exercise
5. Endogenous Mechanisms of Reactive Oxygen and Nitrogen Species Detoxification
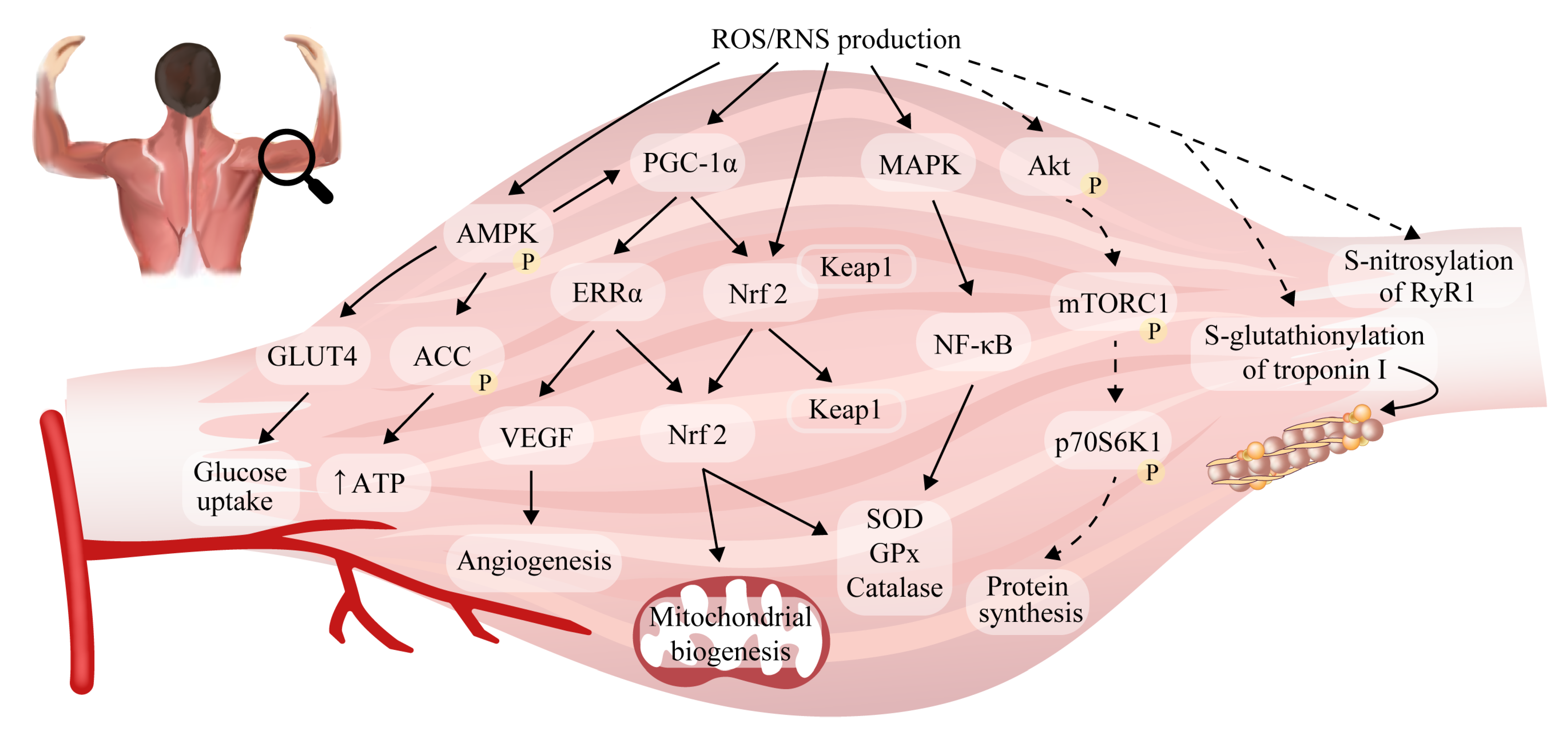
6. Reactive Oxygen/Nitrogen Species and Adaptations to Exercise in Skeletal Muscle
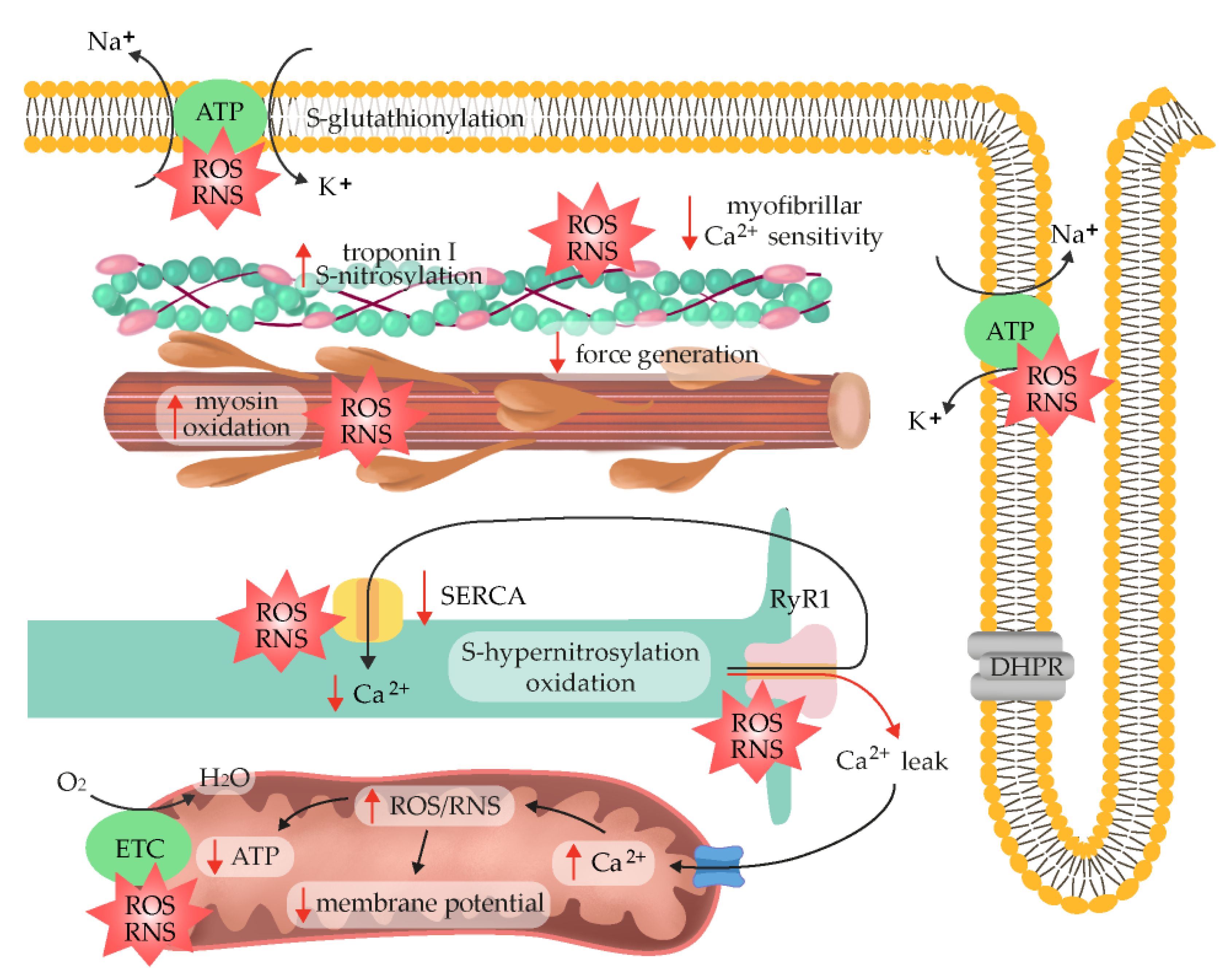
7. ROS/RNS as Fatigue Mediators
8. Exogenous Antioxidants
This entry is adapted from the peer-reviewed paper 10.3390/antiox12020501
References
- Suzuki, K.; Tominaga, T.; Ruhee, R.T.; Ma, S. Characterization and Modulation of Systemic Inflammatory Response to Exhaustive Exercise in Relation to Oxidative Stress. Antioxidants 2020, 9, 401.
- Sahlin, K.; Ren, J.M. Relationship of Contraction Capacity to Metabolic Changes during Recovery from a Fatiguing Contraction. J. Appl. Physiol. 1989, 67, 648–654.
- Woodward, M.; Debold, E.P. Acidosis and Phosphate Directly Reduce Myosin’s Force-Generating Capacity through Distinct Molecular Mechanisms. Front. Physiol. 2018, 9, 862.
- Andrade, F.H.; Reid, M.B.; Westerblad, H. Contractile Response to Low Peroxide Concentrations: Myofibrillar Calcium Sensitivity as a Likely Target for Redox-modulation of Skeletal Muscle Function. FASEB J. 2001, 15, 309–311.
- Christiansen, D.; Eibye, K.; Hostrup, M.; Bangsbo, J. Training with Blood Flow Restriction Increases Femoral Artery Diameter and Thigh Oxygen Delivery during Knee-Extensor Exercise in Recreationally Trained Men. J. Physiol. 2020, 598, 2337–2353.
- Christiansen, D. Molecular Stressors Underlying Exercise Training-Induced Improvements in K + Regulation during Exercise and Na +, K + -ATPase Adaptation in Human Skeletal Muscle. Acta Physiol. 2019, 225, e13196.
- Centner, C.; Zdzieblik, D.; Dressler, P.; Fink, B.; Gollhofer, A.; König, D. Acute Effects of Blood Flow Restriction on Exercise-Induced Free Radical Production in Young and Healthy Subjects. Free Radic. Res. 2018, 52, 446–454.
- Zuo, L.; Clanton, T.L. Reactive Oxygen Species Formation in the Transition to Hypoxia in Skeletal Muscle. Am. J. Physiol.-Cell Physiol. 2005, 289, 207–216.
- Korkmaz, E.; Dönmez, G.; Uzuner, K.; Babayeva, N.; Torgutalp, Ş.Ş.; Özçakar, L. Effects of Blood Flow Restriction Training on Muscle Strength and Architecture. J. Strength Cond. Res. 2022, 36, 1396–1403.
- Anderson, E.J.; Neufer, P.D. Type II Skeletal Myofibers Possess Unique Properties That Potentiate Mitochondrial H2O2 Generation. Am. J. Physiol.-Cell Physiol. 2006, 290, C844–C851.
- Gejl, K.D.; Hvid, L.G.; Andersson, E.P.; Jensen, R.; Holmberg, H.C.; Ørtenblad, N. Contractile Properties of MHC I and II Fibers From Highly Trained Arm and Leg Muscles of Cross-Country Skiers. Front. Physiol. 2021, 12, 855.
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian J. Clin. Biochem. 2015, 30, 11–26.
- Davies, K.J.A.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free Radicals and Tissue Damage Produced by Exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205.
- Dillard, C.J.; Litov, R.E.; Savin, W.M.; Dumelin, E.E.; Tappel, A.L. Effects of Exercise, Vitamin E, and Ozone on Pulmonary Function and Lipid Peroxidation. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1978, 45, 927–932.
- Bailey, D.A.; Lawrenson, L.; McEneny, J.; Young, I.S.; James, P.E.; Jackson, S.K.; Henry, R.R.; Mathieu-Costello, O.; McCord, J.M.; Richardson, R.S. Electron Paramagnetic Spectroscopic Evidence of Exercise-Induced Free Radical Accumulation in Human Skeletal Muscle. Free Radic. Res. 2007, 41, 182–190.
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-Induced Oxidative Stress: Friend or Foe? J. Sport Health Sci. 2020, 9, 415–425.
- Fogarty, M.C.; Hughes, C.M.; Burke, G.; Brown, J.C.; Trinick, T.R.; Duly, E.; Bailey, D.M.; Davison, G.W. Exercise-Induced Lipid Peroxidation: Implications for Deoxyribonucleic Acid Damage and Systemic Free Radical Generation. Environ. Mol. Mutagen. 2011, 52, 35–42.
- Silva, L.A.; Tromm, C.B.; Doyenart, R.; Thirupathi, A.; Silveira, P.C.L.; Pinho, R.A. Effects of Different Frequencies of Physical Training on Electron Transport Chain and Oxidative Damage in Healthy Mice. Motriz. Rev. Educ. Fis. 2018, 24, 101804.
- McNeil, C.J.; Allen, M.D.; Olympico, E.; Shoemaker, J.K.; Rice, C.L. Blood Flow and Muscle Oxygenation during Low, Moderate, and Maximal Sustained Isometric Contractions. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2015, 309, R475–R481.
- Christiansen, D.; Eibye, K.H.; Hostrup, M.; Bangsbo, J. Blood Flow-Restricted Training Enhances Thigh Glucose Uptake during Exercise and Muscle Antioxidant Function in Humans. Metabolism 2019, 98, 1–15.
- Van Der Poel, C.; Edwards, J.N.; MacDonald, W.A.; Stephenson, D.G. Effect of Temperature-Induced Reactive Oxygen Species Production on Excitation-Contraction Coupling in Mammalian Skeletal Muscle. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1482–1487.
- Supruniuk, E.; Maciejczyk, M.; Zalewska, A.; Górski, J.; Chabowski, A. Blood Profile of Cytokines, Chemokines, Growth Factors, and Redox Biomarkers in Response to Different Protocols of Treadmill Running in Rats. Int. J. Mol. Sci. 2020, 21, 8071.
- Bouviere, J.; Fortunato, R.S.; Dupuy, C.; Werneck-De-castro, J.P.; Carvalho, D.P.; Louzada, R.A. Exercise-Stimulated Ros Sensitive Signaling Pathways in Skeletal Muscle. Antioxidants 2021, 10, 537.
- Menazza, S.; Blaauw, B.; Tiepolo, T.; Toniolo, L.; Braghetta, P.; Spolaore, B.; Reggiani, C.; di Lisa, F.; Bonaldo, P.; Canton, M. Oxidative Stress by Monoamine Oxidases Is Causally Involved in Myofiber Damage in Muscular Dystrophy. Hum. Mol. Genet. 2010, 19, 4207–4215.
- Picard, M.; Hepple, R.T.; Burelle, Y. Mitochondrial Functional Specialization in Glycolytic and Oxidative Muscle Fibers: Tailoring the Organelle for Optimal Function. Am. J. Physiol.-Cell Physiol. 2012, 302, 629–641.
- Goncalves, R.L.S.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of Superoxide and Hydrogen Peroxide Production by Muscle Mitochondria Assessed Ex Vivo under Conditions Mimicking Rest and Exercise. J. Biol. Chem. 2015, 290, 209–227.
- Granatiero, V.; Gherardi, G.; Vianello, M.; Salerno, E.; Zecchini, E.; Toniolo, L.; Pallafacchina, G.; Murgia, M.; Blaauw, B.; Rizzuto, R.; et al. Role of P66shc in Skeletal Muscle Function. Sci. Rep. 2017, 7, 6283.
- Alves, J.O.; Pereira, L.M.; Monteiro, I.C.C.D.R.; Dos Santos, L.H.P.; Ferraz, A.S.M.; Loureiro, A.C.C.; Lima, C.C.; Leal-Cardoso, J.H.; Carvalho, D.P.; Fortunato, R.S.; et al. Strenuous Acute Exercise Induces Slow and Fast Twitch-Dependent NADPH Oxidase Expression in Rat Skeletal Muscle. Antioxidants 2020, 9, 57.
- Henríquez-Olguin, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; et al. Cytosolic ROS Production by NADPH Oxidase 2 Regulates Muscle Glucose Uptake during Exercise. Nat. Commun. 2019, 10, 4623.
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of Mitochondrial and Nonmitochondrial Sources Implicate Nicotinamide Adenine Dinucleotide Phosphate Oxidase(s) in the Increased Skeletal Muscle Superoxide Generation That Occurs during Contractile Activity. Antioxid. Redox Signal. 2013, 18, 603–621.
- Javeshghani, D.; Magder, S.A.; Barreiro, E.; Quinn, M.T.; Hussain, S.N.A. Molecular Characterization of a Superoxide-Generating NAD(P)H Oxidase in the Ventilatory Muscles. Am. J. Respir. Crit. Care Med. 2002, 165, 412–418.
- Henríquez-Olguín, C.; Renani, L.B.; Arab-Ceschia, L.; Raun, S.H.; Bhatia, A.; Li, Z.; Knudsen, J.R.; Holmdahl, R.; Jensen, T.E. Adaptations to High-Intensity Interval Training in Skeletal Muscle Require NADPH Oxidase 2. Redox Biol. 2019, 24, 101188.
- Wojtovich, A.P.; Berry, B.J.; Galkin, A. Redox Signaling through Compartmentalization of Reactive Oxygen Species: Implications for Health and Disease. Antioxid. Redox Signal. 2019, 31, 591–593.
- Specht, K.S.; Kant, S.; Addington, A.K.; McMillan, R.P.; Hulver, M.W.; Learnard, H.; Campbell, M.; Donnelly, S.R.; Caliz, A.D.; Pei, Y.; et al. Nox4 Mediates Skeletal Muscle Metabolic Responses to Exercise. Mol. Metab. 2021, 45, 101160.
- Gong, M.C.; Arbogast, S.; Guo, Z.; Mathenia, J.; Su, W.; Reid, M.B. Calcium-Independent Phospholipase A2 Modulates Cytosolic Oxidant Activity and Contractile Function in Murine Skeletal Muscle Cells. J. Appl. Physiol. 2006, 100, 399–405.
- Nethery, D.; Stofan, D.; Callahan, L.; DiMarco, A.; Supinski, G. Formation of Reactive Oxygen Species by the Contracting Diaphragm Is PLA2 Dependent. J. Appl. Physiol. 1999, 87, 792–800.
- Zhao, X.; Bey, E.A.; Wientjes, F.B.; Cathcart, M.K. Cytosolic Phospholipase A2 (CPLA2) Regulation of Human Monocyte NADPH Oxidase Activity: CPLA2 Affects Translocation but Not Phosphorylation of P67phox and P47phox. J. Biol. Chem. 2002, 277, 25385–25392.
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Bao, S.; Clanton, T.L. Lipoxygenase-Dependent Superoxide Release in Skeletal Muscle. J. Appl. Physiol. 2004, 97, 661–668.
- Gomez-Cabrera, M.C.; Borrás, C.; Pallardo, F.V.; Sastre, J.; Ji, L.L.; Viña, J. Decreasing Xanthine Oxidase-Mediated Oxidative Stress Prevents Useful Cellular Adaptations to Exercise in Rats. J. Physiol. 2005, 567, 113–120.
- Sutkowy, P.; Wróblewska, J.; Wróblewski, M.; Nuszkiewicz, J.; Modrzejewska, M.; Woźniak, A. The Impact of Exercise on Redox Equilibrium in Cardiovascular Diseases. J. Clin. Med. 2022, 11, 4833.
- Tidball, J.G.; Wehling-Henricks, M. Nitric Oxide Synthase Deficiency and the Pathophysiology of Muscular Dystrophy. J. Physiol. 2014, 592, 4627–4638.
- Joyner, M.J.; Coyle, E.F. Endurance Exercise Performance: The Physiology of Champions. J. Physiol. 2008, 586, 35–44.
- Cheng, A.J.; Yamada, T.; Rassier, D.E.; Andersson, D.C.; Westerblad, H.; Lanner, J.T. Reactive Oxygen/Nitrogen Species and Contractile Function in Skeletal Muscle during Fatigue and Recovery. J. Physiol. 2016, 594, 5149–5160.
- Kawamura, T.; Muraoka, I. Exercise-Induced Oxidative Stress and the Effects of Antioxidant Intake from a Physiological Viewpoint. Antioxidants 2018, 7, 119.
- Beckman, J.S.; Koppenol, W.H. Nitric Oxide, Superoxide, and Peroxynitrite: The Good, the Bad, and the Ugly. Am. J. Physiol.-Cell Physiol. 1996, 271, C1424–C1437.
- Powers, S.K.; Jackson, M.J. Exercise-Induced Oxidative Stress: Cellular Mechanisms and Impact on Muscle Force Production. Physiol. Rev. 2008, 88, 1243–1276.
- Wiecek, M.; Maciejczyk, M.; Szymura, J.; Kantorowicz, M.; Szygula, Z. Impact of Single Anaerobic Exercise on Delayed Activation of Endothelial Xanthine Oxidase in Men and Women. Redox Rep. 2017, 22, 367–376.
- Alessio, H.M.; Hagerman, A.E.; Fulkerson, B.K.; Ambrose, J.; Rice, R.E.; Wiley, R.L. Generation of Reactive Oxygen Species after Exhaustive Aerobic and Isometric Exercise. Med. Sci. Sport. Exerc. 2000, 32, 1576–1581.
- Groussard, C.; Rannou-Bekono, F.; Machefer, G.; Chevanne, M.; Vincent, S.; Sergent, O.; Cillard, J.; Gratas-Delamarche, A. Changes in Blood Lipid Peroxidation Markers and Antioxidants after a Single Sprint Anaerobic Exercise. Eur. J. Appl. Physiol. 2003, 89, 14–20.
- He, F.; Li, J.; Liu, Z.; Chuang, C.C.; Yang, W.; Zuo, L. Redox Mechanism of Reactive Oxygen Species in Exercise. Front. Physiol. 2016, 7, e0185993.
- Morales-Alamo, D.; Calbet, J.A.L. Free Radicals and Sprint Exercise in Humans. Free Radic. Res. 2014, 48, 30–42.
- Radak, Z.; Asano, K.; Inoue, M.; Kizaki, T.; Oh-Ishi, S.; Suzuki, K.; Taniguchi, N.; Ohno, H. Superoxide Dismutase Derivative Reduces Oxidative Damage in Skeletal Muscle of Rats during Exhaustive Exercise. J. Appl. Physiol. 1995, 79, 129–135.
- Sloboda, D.D.; Brooks, S.V. Reactive Oxygen Species Generation Is Not Different during Isometric and Lengthening Contractions of Mouse Muscle. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2013, 305, R832.
- Pye, D.; Palomero, J.; Kabayo, T.; Jackson, M.J. Real-Time Measurement of Nitric Oxide in Single Mature Mouse Skeletal Muscle Fibres during Contractions. J. Physiol. 2007, 581, 309–318.
- Steinbacher, P.; Eckl, P. Impact of Oxidative Stress on Exercising Skeletal Muscle. Biomolecules 2015, 5, 356–377.
- Vincent, H.K.; Powers, S.K.; Stewart, D.J.; Demirel, H.A.; Shanely, R.A.; Naito, H. Short-Term Exercise Training Improves Diaphragm Antioxidant Capacity and Endurance. Eur. J. Appl. Physiol. Occup. Physiol. 2000, 81, 67–74.
- Vincent, H.K.; Powers, S.K.; Demirel, H.A.; Coombes, J.S.; Naito, H. Exercise Training Protects against Contraction-Induced Lipid Peroxidation in the Diaphragm. Eur. J. Appl. Physiol. Occup. Physiol. 1999, 79, 268–273.
- García, J.J.; Berzosa, C.; Cebrián, I.; Fuentes-Broto, L.; Gómez-Trullén, E.; Piedrafita, E.; Martínez-Ballarín, E.; López-Pingarrn, L.; Reiter, R.J. Acute Exercise Increases Plasma Total Antioxidant Status and Antioxidant Enzyme Activities in Untrained Men. J. Biomed. Biotechnol. 2011, 2011, 540458.
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-Induced Skeletal Muscle Remodeling and Metabolic Adaptation: Redox Signaling and Role of Autophagy. Antioxid. Redox Signal. 2014, 21, 154–176.
- Larkin, L.M.; Davis, C.S.; Sims-Robinson, C.; Kostrominova, T.Y.; van Remmen, H.; Richardson, A.; Feldman, E.L.; Brooks, S.V. Skeletal Muscle Weakness Due to Deficiency of CuZn-Superoxide Dismutase Is Associated with Loss of Functional Innervation. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 301, R1400–R1407.
- Sakellariou, G.K.; McDonagh, B.; Porter, H.; Giakoumaki, I.I.; Earl, K.E.; Nye, G.A.; Vasilaki, A.; Brooks, S.V.; Richardson, A.; Van Remmen, H.; et al. Comparison of Whole Body SOD1 Knockout with Muscle-Specific SOD1 Knockout Mice Reveals a Role for Nerve Redox Signaling in Regulation of Degenerative Pathways in Skeletal Muscle. Antioxid. Redox Signal. 2018, 28, 275–295.
- Kuwahara, H.; Horie, T.; Ishikawa, S.; Tsuda, C.; Kawakami, S.; Noda, Y.; Kaneko, T.; Tahara, S.; Tachibana, T.; Okabe, M.; et al. Oxidative Stress in Skeletal Muscle Causes Severe Disturbance of Exercise Activity without Muscle Atrophy. Free Radic. Biol. Med. 2010, 48, 1252–1262.
- Radak, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen Consumption and Usage during Physical Exercise: The Balance between Oxidative Stress and ROS-Dependent Adaptive Signaling. Antioxid. Redox Signal. 2013, 18, 1208–1246.
- Pengam, M.; Amérand, A.; Simon, B.; Guernec, A.; Inizan, M.; Moisan, C. How Do Exercise Training Variables Stimulate Processes Related to Mitochondrial Biogenesis in Slow and Fast Trout Muscle Fibres? Exp. Physiol. 2021, 106, 938–957.
- MacInnis, M.J.; Gibala, M.J. Physiological Adaptations to Interval Training and the Role of Exercise Intensity. J. Physiol. 2017, 595, 2915.
- Bishop, D.J.; Botella, J.; Granata, C. CrossTalk Opposing View: Exercise Training Volume Is More Important than Training Intensity to Promote Increases in Mitochondrial Content. J. Physiol. 2019, 597, 4115–4118.
- Novelli, G.P.; Bracciotti, G.; Falsini, S. Spin-Trappers and Vitamin E Prolong Endurance to Muscle Fatigue in Mice. Free Radic. Biol. Med. 1990, 8, 9–13.
- Lamb, G.D.; Posterino, G.S. Effects of Oxidation and Reduction on Contractile Function in Skeletal Muscle Fibres of the Rat. J. Physiol. 2003, 546, 149–163.
- Nogueira, L.; Figueiredo-Freitas, C.; Casimiro-Lopes, G.; Magdesian, M.H.; Assreuy, J.; Sorenson, M.M. Myosin Is Reversibly Inhibited by S-Nitrosylation. Biochem. J. 2009, 424, 221–231.
- Dutka, T.L.; Mollica, J.P.; Lamb, G.D. Differential Effects of Peroxynitrite on Contractile Protein Properties in Fast- and Slow-Twitch Skeletal Muscle Fibers of Rat. J. Appl. Physiol. 2011, 110, 705–716.
- Ji, L.L.; Fu, R.; Mitchell, E.W. Glutathione and Antioxidant Enzymes in Skeletal Muscle: Effects of Fiber Type and Exercise Intensity. J. Appl. Physiol. 1992, 73, 1854–1859.
- Fauler, M.; Jurkat-Rott, K.; Lehmann-Horn, F. Membrane Excitability and Excitation-Contraction Uncoupling in Muscle Fatigue. Neuromuscul. Disord. 2012, 22, S162–S167.
- Watanabe, D.; Wada, M.; Hogan, M.; Hepple, R.; Watanabe, D.; Wada, M. Fatigue-Induced Change in T-System Excitability and Its Major Cause in Rat Fast-Twitch Skeletal Muscle In Vivo. J. Physiol. 2020, 598, 5195–5211.
- Moon, Y.; Balke, J.E.; Madorma, D.; Siegel, M.P.; Knowels, G.; Brouckaert, P.; Buys, E.S.; Marcinek, D.J.; Percival, J.M. Nitric Oxide Regulates Skeletal Muscle Fatigue, Fiber Type, Microtubule Organization, and Mitochondrial ATP Synthesis Efficiency Through CGMP-Dependent Mechanisms. Antioxid. Redox Signal. 2017, 26, 966–985.
- McKenna, M.J.; Medved, I.; Goodman, C.A.; Brown, M.J.; Bjorksten, A.R.; Murphy, K.T.; Petersen, A.C.; Sostaric, S.; Gong, X. N-Acetylcysteine Attenuates the Decline in Muscle Na+, K+-Pump Activity and Delays Fatigue during Prolonged Exercise in Humans. J. Physiol. 2006, 576, 279–288.
- Place, N.; Yamada, T.; Zhang, S.J.; Westerblad, H.; Bruton, J.D. High Temperature Does Not Alter Fatigability in Intact Mouse Skeletal Muscle Fibres. J. Physiol. 2009, 587, 4717–4724.
- Radak, Z.; Ishihara, K.; Tekus, E.; Varga, C.; Posa, A.; Balogh, L.; Boldogh, I.; Koltai, E. Exercise, Oxidants, and Antioxidants Change the Shape of the Bell-Shaped Hormesis Curve. Redox Biol. 2017, 12, 285–290.
- Wu, M.; Zhao, A.; Yan, X.; Gao, H.; Zhang, C.; Liu, X.; Luo, Q.; Xie, F.; Liu, S.; Shi, D. Hepatic AMPK Signaling Activation in Response to Dynamic REDOX Balance Is a Biomarker of Exercise to Improve Blood Glucose Control. eLife 2022, 11, e79939.
- Gomez-Cabrera, M.C.; Domenech, E.; Viña, J. Moderate Exercise Is an Antioxidant: Upregulation of Antioxidant Genes by Training. Free Radic. Biol. Med. 2008, 44, 126–131.
- Trapp, D.; Knez, W.; Sinclair, W. Could a Vegetarian Diet Reduce Exercise-Induced Oxidative Stress? A Review of the Literature. J. Sport. Sci. 2010, 28, 1261–1268.
- Peternelj, T.T.; Coombes, J.S. Antioxidant Supplementation during Exercise Training: Beneficial or Detrimental? Sport. Med. 2011, 41, 1043–1069.
- Nieman, D.C.; Henson, D.A.; McAnulty, S.R.; McAnulty, L.S.; Morrow, J.D.; Ahmed, A.; Heward, C.B. Vitamin E and Immunity after the Kona Triathlon World Championship. Med. Sci. Sport. Exerc. 2004, 36, 1328–1335.
- Sacheck, J.M.; Milbury, P.E.; Cannon, J.G.; Roubenoff, R.; Blumberg, J.B. Effect of Vitamin E and Eccentric Exercise on Selected Biomarkers of Oxidative Stress in Young and Elderly Men. Free Radic. Biol. Med. 2003, 34, 1575–1588.
- Close, G.L.; Ashton, T.; Cable, T.; Doran, D.; Holloway, C.; McArdle, F.; MacLaren, D.P.M. Ascorbic Acid Supplementation Does Not Attenuate Post-Exercise Muscle Soreness Following Muscle-Damaging Exercise but May Delay the Recovery Process. Br. J. Nutr. 2006, 95, 976–981.
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants Prevent Health-Promoting Effects of Physical Exercise in Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670.
- Paulsen, G.; Cumming, K.T.; Holden, G.; Hallén, J.; Rønnestad, B.R.; Sveen, O.; Skaug, A.; Paur, I.; Bastani, N.E.; Østgaard, H.N.; et al. Vitamin C and E Supplementation Hampers Cellular Adaptation to Endurance Training in Humans: A Double-Blind, Randomised, Controlled Trial. J. Physiol. 2014, 592, 1887–1901.
- Makanae, Y.; Kawada, S.; Sasaki, K.; Nakazato, K.; Ishii, N. Vitamin C Administration Attenuates Overload-Induced Skeletal Muscle Hypertrophy in Rats. Acta Physiol. 2013, 208, 57–65.
- Theodorou, A.A.; Nikolaidis, M.G.; Paschalis, V.; Koutsias, S.; Panayiotou, G.; Fatouros, I.G.; Koutedakis, Y.; Jamurtas, A.Z. No Effect of Antioxidant Supplementation on Muscle Performance and Blood Redox Status Adaptations to Eccentric Training. Am. J. Clin. Nutr. 2011, 93, 1373–1383.
- Richardson, R.S.; Donato, A.J.; Uberoi, A.; Wray, D.W.; Lawrenson, L.; Nishiyama, S.; Bailey, D.M. Exercise-Induced Brachial Artery Vasodilation: Role of Free Radicals. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H1516–H1522.
- Wray, D.W.; Uberoi, A.; Lawrenson, L.; Bailey, D.M.; Richardson, R.S. Oral Antioxidants and Cardiovascular Health in the Exercise-Trained and Untrained Elderly: A Radically Different Outcome. Clin. Sci. 2009, 116, 433–441.
- Sindler, A.L.; Delp, M.D.; Reyes, R.; Wu, G.; Muller-Delp, J.M. Effects of Ageing and Exercise Training on ENOS Uncoupling in Skeletal Muscle Resistance Arterioles. J. Physiol. 2009, 587, 3885–3897.
- Rochette, L.; Ghibu, S.; Richard, C.; Zeller, M.; Cottin, Y.; Vergely, C. Direct and Indirect Antioxidant Properties of α-Lipoic Acid and Therapeutic Potential. Mol. Nutr. Food Res. 2013, 57, 114–125.
- Henriksen, E.J. Exercise Training and the Antioxidant α-Lipoic Acid in the Treatment of Insulin Resistance and Type 2 Diabetes. Free Radic. Biol. Med. 2006, 40, 3–12.
- Maciejczyk, M.; Żebrowska, E.; Nesterowicz, M.; Supruniuk, E.; Choromańska, B.; Chabowski, A.; Żendzian-Piotrowska, M.; Zalewska, A. α-Lipoic Acid Reduces Ceramide Synthesis and Neuroinflammation in the Hypothalamus of Insulin-Resistant Rats, While in the Cerebral Cortex Diminishes the β-Amyloid Accumulation. J. Inflamm. Res. 2022, 15, 2295–2312.
- Pingitore, A.; Lima, G.P.P.; Mastorci, F.; Quinones, A.; Iervasi, G.; Vassalle, C. Exercise and Oxidative Stress: Potential Effects of Antioxidant Dietary Strategies in Sports. Nutrition 2015, 31, 916–922.
- Isenmann, E.; Trittel, L.; Diel, P. The Effects of Alpha Lipoic Acid on Muscle Strength Recovery after a Single and a Short-Term Chronic Supplementation—A Study in Healthy Well-Trained Individuals after Intensive Resistance and Endurance Training. J. Int. Soc. Sport. Nutr. 2020, 17, 1–13.
- Coombes, J.S.; Powers, S.K.; Rowell, B.; Hamilton, K.L.; Dodd, S.L.; Shanely, R.A.; Sen, C.K.; Packer, L. Effects of Vitamin E and α-Lipoic Acid on Skeletal Muscle Contractile Properties. J. Appl. Physiol. 2001, 90, 1424–1430.