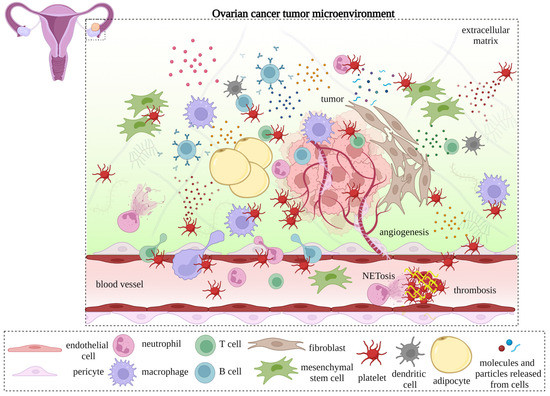
Healthy individuals have platelet counts of between 150,000 and 450,000 per microliter of blood. In contrast, roughly one-third of newly diagnosed ovarian cancer patients have platelet counts exceeding 450,000 per microliter [
27]. In patients with ovarian cancer, thrombocytosis is an adverse prognostic factor associated with elevated serum carcinoma antigen 125 (CA-125) levels, advanced disease stage, and poor clinical outcomes [
27,
28], as well as the diminished efficacy of secondary cytoreductive surgery [
29] and chemotherapy [
30]. Aside from contributing to the formation of venous thromboembolisms, platelets contribute to cancer progression via distinct mechanisms, including increasing proliferation [
31], epithelial–mesenchymal transition (EMT) [
32], and anoikis resistance in cancer cells [
33]; promoting the formation of the premetastatic niche and metastasis [
33]; enhancing angiogenesis [
27] and the integrity of tumor vasculature [
34]; inducing immune tolerance [
35]; and reducing the impact of chemotherapy [
30].
Table 1. The major components reside on activated platelets or are released from platelet granules.
2. Interactions of Platelets with TME Compartments: Endothelial Cells, Pericytes, and Cancer-Associated Fibroblasts
2.1. Interactions with Endothelial Cells
2.1.1. In Angiogenesis
Tumor angiogenesis involves degradation of the vascular endothelial matrix, the proliferation and migration of endothelial cells, the branching of endothelial cells to generate vascular rings, and the establishment of new basement membranes [
81]. Tumor blood vessels, which tend to be erratic, branched, and leaky, are dissimilar to normal blood vessels in terms of shape, integrity, and permeability. Moreover, perivascular cells are reduced in number and are less likely to be associated with endothelial cells [
82,
83]. Blood flow in tumor-associated vessels is inconsistent and may lead to maladjusted circulation [
82,
84]. Consequently, tumors cannot receive adequate oxygen and nutrients, and discharge excess carbon dioxide and other metabolites generated by the glycolytic pathway. The TME becomes more hypoxic, acidic, and ischemic [
85]. In addition, the hyperpermeability of the tumor vasculature enhances extravascular clotting, fibrin gel clot formation, and endothelial and stromal cell expansion [
86]. Angiogenesis is a poor prognostic factor in ovarian cancer [
87], and antiangiogenic therapeutics demonstrate a moderate effect on overall and progression-free survival [
88,
89].
Platelets preferentially attach to tumor-associated vessels rather than normal vasculature, amplifying the delivery of tumorigenic mediators to the TME [
90]. Tumor cell-induced platelet activation (TCIPA) leads to the translocation of P-selectin (also known as CD62P), a cell adhesion molecule stored in the α-granules [
37], to the platelet surface. The binding of P-selectin to the P-selectin glycoprotein ligand (PSGL-1) on leukocytes governs leukocyte rolling in activated endothelial cells [
91] and the generation of platelet—cancer cell complexes [
92]. Adhesion molecules, including integrins, von Willebrand factor (vWF), fibrinogen, fibronectin, and coagulation factors, and several members of the a disintegrin and metalloproteinase (ADAM) protein family accommodate the activation, tethering, rolling, and firm adhesion of platelets to endothelial cells [
93]. Activated platelets degranulate and release various factors that affect angiogenesis. More than 30 components associated with platelets that influence angiogenesis have been described [
94]. Platelets generate angiostatic factors such as endostatin, angiostatin, and thrombospondin-1 (TSP-1), and angiogenic factors including vascular endothelial growth factor (VEGF), angiopoietin-1, stromal-derived factor 1 (SDF-1, also known as the chemokine (C-X-C motif) ligand [CXCL]12), sphingosine 1-phosphate (S1P), transforming growth factor-beta (TGF-β), interleukin (IL)-6, and platelet factor 4 (PF4; also known as CXCL4) [
61]. Platelet-derived growth factor (PDGF) supports the function of cancer-associated fibroblasts (CAFs), vascular pericytes, and smooth muscle cells in angiogenesis [
95]. Platelets also support the recruitment of endothelial progenitor cells (EPCs) [
96]. Platelet integrin GPIIb/IIIa promotes endothelial cell proliferation and function [
97]. The activation of platelets and the release of their granular content, such as angiopoietin-1 and serotonin, prevent intratumoral bleeding [
98]. ATP released from the δ-granules of platelets activates endothelial P2Y
2 receptors, causing the retraction of endothelial cells and promoting the transendothelial migration of cancer cells (intravasation and extravasation) and metastasis [
99]. Platelet microparticles increase the expression of matrix metalloproteinases (MMPs) on endothelial cells [
100], assisting in the generation of new vessels [
61].
The co-localization of GPIIb (CD41), platelet endothelial cell adhesion molecule-1 (PECAM-1; also known as CD31), and VEGF in ovarian cancer tissues suggests the involvement of platelets in angiogenesis and tumor growth [
101]. An increased level of VEGF can be considered a biomarker of ovarian cancer [
102] and an indicator of advanced disease, ascites formation, metastasis, and reduced survival [
103,
104]. Moreover, the levels of PDGF-BB and VEGF were found to be positively correlated in the TME and ascites, and the pharmacological inhibition of their receptors increased the efficacy of chemotherapy in patients with ovarian cancer [
105]. The co-localization of regulator of G-protein signaling 5 (RGS5), a signal transduction molecule upregulated in endothelial cells in the TME, with PECAM-1 and PDGF receptor (PDGFR)-β, has been reported in various types of cancer, including ovarian cancer [
106]. The participation of activated platelets in angiogenesis is displayed in
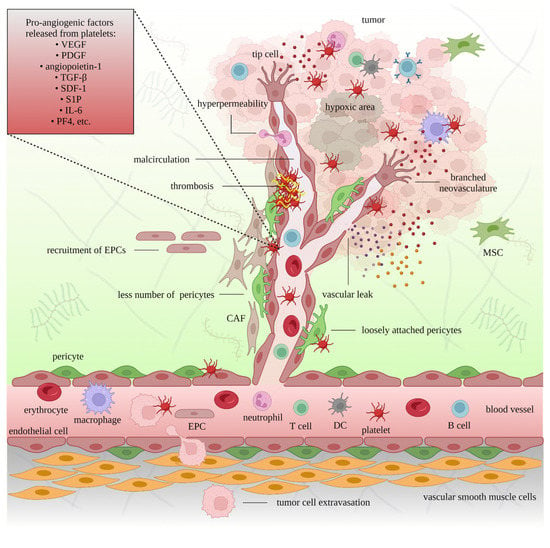
Figure 2.
Figure 2. The pro-angiogenic role of platelets in cancer. The interaction between platelets, endothelial cells, and pericytes supports the extravasation of immune cells, mesenchymal stem cells (MSCs), endothelial precursor cells (EPCs), and tumor cells. Platelets also release proangiogenic factors that promote new blood vessel formation and facilitate tumor growth. The newly formed cancer-associated blood vessels are branched, leaky, and less supported by pericytes. Insufficient oxygen and nutrient supplies lead to hypoxic and necrotic areas in the tumor. Platelet-targeting strategies can restrict neoangiogenesis.
Platelet GPIb-IX receptor complex, a receptor for vWF, and GPIIb/IIIa, the receptor for fibrinogen, promote platelet aggregation [
107] and adhesion to endothelial cells. Additionally, platelet P-selectin and GPIIb assist in the adhesion of platelets to cancer cells. Hence, platelets assist in the clinging of cancer cells to the endothelium and metastasis [
108,
109]. Targeting platelet surface proteins might show therapeutic benefits in cancer. Antiplatelet agent-directed platelet inhibition diminishes the proliferative capability of ovarian cancer cells [
31]. In addition, focal adhesion kinase (FAK) promotes platelet infiltration into the TME, and targeting FAK suppresses ovarian tumor growth. Dual therapy using antiplatelet agents and antiangiogenic drugs prevents rebound tumor growth after discontinuing antiangiogenic agents [
20].
2.1.2. In Lymphangiogenesis
Lymphangiogenesis is the formation of new lymphatic vessels and occurs during embryonic development and in pathological conditions involving inflammation and tumor metastasis [
110]. Platelets are essential for the proper partitioning of blood and lymphatic vessels during development, mainly by coordinating endothelial cells’ expansion, relocation, and tube formation. This phenomenon occurs following the engagement of platelet CLEC-2 with its ligand podoplanin on lymphatic endothelial cells [
111]. Although platelets do not necessarily contribute to the maintenance of the separation of the two circulatory systems post-development in normal conditions, in certain situations, including wound healing or tumor growth, platelets again participate in lymphangiogenesis [
112]. Platelets stimulate lymphangiogenesis by secreting proangiogenic factors such as VEGF, angiopoietin-1, PDGF, and insulin-like growth factor 1 (IGF-1) [
61,
113], and through the interaction of CLEC-2 and podoplanin [
111].
In patients with ovarian cancer, the upregulation of lymphangiogenic markers is associated with more aggressive disease and shorter overall survival [
114]. Podoplanin overexpression in the malignant stroma of ovarian cancer patients predicts lymphatic spread and poor clinical outcomes [
115]. Blocking podoplanin—CLEC-2 contact between ovarian cancer cells and platelets forestalls lymph vessel proliferation [
116]. Likewise, VEGF and PDGF released by platelets promote lymph vessel generation in epithelial ovarian cancer [
117]. Treatment with antiangiogenic agents might attenuate lymphangiogenesis in ovarian cancer [
118]. Notably, inhibiting the TGF-β signaling cascade prevents lymphangiogenesis and subsequent VEGF-mediated ascites generation in ovarian cancer patients [
119].
2.2. Interactions with Pericytes
Pericytes are perivascular cells embedded in the basement membrane surrounding the microvasculature [
120]. Pericytes might prevent the intravasation of cancer cells and metastasis [
121]; however, they might also facilitate micrometastasis by supporting the formation of tumor vasculature [
122]. Tumor vessels have atypical coverage of pericytes, whose contact with endothelial cells is disrupted [
122]. The tumor vasculature has an excess of pericytes that loosely interact with endothelial cells, deteriorating the integrity of the vessels and resulting in hemorrhage [
123]. In ovarian cancer, abnormal pericyte numbers and expression signatures are associated with tumor growth, aggressive metastasis, and poor clinical outcomes [
124].
Podoplanin, which is highly expressed on pericytes, mediates platelet binding to pericytes via CLEC-2 [
125]. Furthermore, platelet-derived TGF-β, angiopoietin, and PDGF also stimulate pericyte differentiation, colonization, and their interaction with endothelial cells [
64]. TGF-β strongly influences the proliferation of pericytes [
126] and their association with endothelial cells through the TGF-β—matrix protein axis [
127]. The activation of the TGF-β signaling cascade impacts the density and lumen size of tumor microvessels [
127]. Angiopoietin overexpression is linked with pericyte impairment and tumor vessel instability [
128]. Blocking TGF-β or angiopoietin signaling inhibits tumor growth and neovascularization [
127,
129]. PDGF released from platelets and other cells [
130] is essential for pericytes recruitment and function during tumor neoangiogenesis. Preventing PDGF isoforms from binding to their receptors and hindering angiogenesis with antiangiogenic agents such as bevacizumab may interfere with the incorporation of pericytes into new blood vessels [
131].
TSP-1 is a matricellular glycoprotein with antiangiogenic properties that counters the proliferative effects of growth factors on endothelial cells [
132,
133]. TSP-1 is released from the granules of activated platelets, prompting platelet aggregation and tethering [
56]. In ovarian cancer, binding of the TSP-1 three type 1 repeats (3TSR) domain of TSP-1 to GPIV (CD36) normalizes the tumor vasculature and exhibits antitumor function [
134]. Treating patients with 3TSR in combination with chemotherapeutics [
135] or oncolytic viruses [
136] can improve the efficacy of anticancer therapies. 3TSR alone, or fused with the Fc region of human IgG1 for improved stability, increases the number of pericyte-covered blood vessels, reduces the proliferative capacity of endothelial cells, and contributes to vascular normalization [
134].
Platelets release IL-6 [
73] and also trigger IL-6 secretion from tumor cells by releasing several factors, such as lysophosphatidic acid (LPA) [
137]. The protumorigenic cytokine IL-6 is significantly elevated in ovarian cancer patients with confirmed thrombocytosis [
27]. High IL-6 levels promote neoangiogenesis with abnormal pericyte coating. Anti-VEGF and anti-IL-6 agents reduce vessel sprouting and leakiness of the vasculature by reinstating the pericyte lining [
138]. Combining chemotherapeutic agents and pazopanib, a multitargeted tyrosine kinase inhibitor, can help restore pericyte coverage and restrict tumor microvessel density [
139] in patients with ovarian cancer. The antiplatelet action of pazopanib [
140] might further inhibit tumor growth and angiogenesis [
20].
2.3. Interactions with Cancer-Associated Fibroblasts
Fibroblasts are a heterogeneous population of connective tissue cells with a presumably mesenchymal origin [
141]. Fibroblasts can differentiate into particular mesenchymal cell types, including osteoblasts, adipocytes, and chondrocytes [
142]. In the TME, CAFs promote disease progression by releasing various molecules; rearranging the ECM to facilitate cancer cell motility, invasion, and EMT; and stimulating angiogenesis, tumor growth, and metastasis. CAFs modulate the function of immune cells and the metabolism of cancer cells to promote tumor survival [
143]. CAFs also release extracellular vesicles that support cancer progression [
144] and chemoresistance [
145]. Although fibroblasts play a role in tumorigenesis, they may also restrict tumor development by activating the tumoricidal immune response or consolidating the ECM to prevent tumor dissemination [
146].
Extravasated platelets promote EMT by releasing mediators such as TGF-β, SDF-1, and PDGF. The same mediators also induce the differentiation, migration, and proliferation of CAFs [
147,
148]. PDGF and TGF-β induce mesenchymal stem cell (MSC) differentiation into CAFs [
149]. Integrin α11 is a CAF marker, and its expression is related to myofibroblast differentiation and ECM alteration. CAFs expressing integrin α11 and PDGFR-β are associated with poor clinical outcomes in ovarian cancers and other malignancies [
150]. Platelet-originated CLEC-2 induces the migration and proliferation of CAFs in the TME [
15]. The binding of platelet-derived CLEC-2 to podoplanin on CAFs and cancer cells promote tumor growth and venous thrombosis in patients with ovarian cancer [
125,
151]. LPA derived from ovarian cancer cells promotes the differentiation of fibroblasts into CAFs through a hypoxia-inducible factor 1 alpha (HIF-1α)-dependent mechanism [
152]. LPA activates platelets [
153], which, in turn, release LPA [
137]. Moreover, LPA promotes the secretion of VEGF and SDF-1 from MSCs, further supporting ovarian cancer progression [
154]. Under oxidative stress, platelets release their mitochondria, which are picked up by MSCs [
24]. Mitochondria originating in platelets and engulfed by MSCs promote fatty acid synthesis and ATP production and stimulate the release of angiogenic components, such as VEGF and hepatocyte growth factor (HGF), from MSCs [
24].
CAFs originating from MSCs release platelet-activating factor (PAF), promoting platelet activation and aggregation [
155], which further supports ovarian cancer progression and induces ovarian cancer development through the PAF/PAF receptor signaling pathway [
34]. Exosomes from ovarian cancer cells induce the generation of CAFs from MSCs in the tumor stroma [
156]. CAF-released IL-6 causes EMT in ovarian cancer cells, tumor growth, and ECM reorganization, mainly by mediating STAT3 phosphorylation [
157]. The increased concentrations of IL-6 in the stroma or ascites can activate platelet function and aggregation and lead to thrombosis [
158].
Most patients with metastatic ovarian cancer have peritoneal dissemination, which indicates a poor prognosis. It starts with the emigration of cancer cells into the peritoneal fluid, forming floating masses that attach to peritoneal mesothelial cells throughout the peritoneal cavity [
159]. Alternatively, the cancer cells can initiate an inflammatory reaction in the peritoneal stroma, promoting the generation of a fibrin mesh that can be used for adhesion to the peritoneal surface. Fibrin mesh can also potentiate the colonization of fibroblasts and endothelial cells. Fibroblasts that differentiate into CAFs promote ovarian cancer invasion through the upregulation of several markers such as alpha-smooth muscle actin (α-MA), PDGFR, and podoplanin [
151]. A subset of CAFs originating from mesothelial cells through mesothelial-to-mesenchymal transition (MMT) contribute to peritoneal metastasis [
160]. Ovarian cancer cells that have been relocated to the peritoneal cavity promote ascites, and the ascitic fluid contains various cytokines and growth factors [
161]. TGF-β derived from ascites and activated platelets is one of the main stimulating factors for MMT [
160,
162]. In addition, tissue factor (TF), present in high amounts in ascites, cancer cell masses, and cancer cell-derived microparticles, induces thrombin generation, which activates platelets and produces fibrin [
163]. Activated platelets further increase the expression of TF, prompting ovarian cancer migration [
164]. The activation of mesothelial cells by TGF-β released from platelets is partially responsible for ECM remodeling during metastasis [
165].
Like fibroblasts, mesothelial cells and adipocytes in the omentum and peritoneum can be prompted by cancer cells to differentiate into CAFs [
166]. Periostin is a secretory protein that is overexpressed by stromal fibroblasts in multiple cancers, including ovarian cancer, and its overexpression is associated with poor clinical outcomes in patients with epithelial ovarian cancer. TGF-β modulates periostin expression and promotes ovarian cancer growth and chemotherapy resistance [
167]. Similarly, the aberrant expression and release of connective tissue growth factor (CTGF), a stromal factor, induces the colonization and peritoneal adhesion of ovarian cancer cells [
168]. Platelets store a large quantity of CTGF [
63], suggesting that platelet activation and the release of CTGF may participate in ovarian cancer seeding in the peritoneum.
Inhibitors of the TGF-β signaling pathway can diminish CAFs’ function in ovarian cancer [
169]. Anti-VEGF therapy can inhibit ovarian cancer progression, metastasis, and malignant ascites formation promoted by the release of VEGF from CAFs, along with various other cell types [
170]; however, synchronous anti-PDGF treatment might be necessary to target CAFs resistant to VEGF-neutralizing agents [
171]. It has been reported that aspirin therapy suppresses chemotherapy-induced CAF formation in colorectal cancer [
172] and the impact of CAFs on ovarian cancer.