Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Obesity is a major public health concern associated with chronic low-grade systemic inflammation. Moreover, obesity is considered one of the major risk factors for the development of several chronic diseases, such as cancer. Researchers describe here, how adipose tissue dysfunction, particularly alterations in adipocytes and macrophages, participate in such processes.
- adipose tissue
- obesity
- inflammation
- endocrine signaling
- cancer cells
1. Introduction
1.1. Obesity: A Health Problem
Obesity is characterized by an excessive accumulation of white adipose tissue that elevates health risks. The already epidemic proportions continue to grow throughout the world and are of great concern given that numerous diseases are either triggered or aggravated by this disorder. Indeed, the COVID-19 pandemic revealed that people with obesity were at greater risk for unfavorable outcomes related to disease severity and death [1]. Obesity is a complex multifactorial disease with etiological components that range from molecular to social factors, and given the well-established connection to the development of multiple chronic diseases, it is necessary to better understand the molecular mechanisms involved in order to prevent or revert related comorbidities.
1.2. White Adipose Tissue Dysfunction: Role of Macrophages and Adipocytes
For a long time, adipose tissue was considered a simple energy reservoir; however, after the first reports in the 1990s on its ability to secrete numerous key regulatory factors, this tissue has come to be recognized as a major endocrine organ [2]. Adipose tissue communicates with other organs and maintains homeostasis through the release of many different mediators, including lipids, metabolites, noncoding RNAs, and extracellular vesicles (EVs) [3]. Importantly, obesity is frequently characterized by adipose tissue dysfunction and infiltration by macrophages, ensuing changes that result in the liberation of many proinflammatory factors, insulin resistance, and impaired lipid metabolism. Local changes in adipose tissue are transmitted to various organs via this altered secretome and thereby contribute to deleterious forms of organ crosstalk typically associated with the development of chronic diseases, such as cancer [4].
Adipose tissue expansion due to excessive energy intake is accompanied by numerous changes in tissue function and composition. Particularly, dysfunctional fat is characterized by inflammation, driven mainly by the infiltration of proinflammatory CD11c-expressing M1-polarized macrophages. Once the tissue has been infiltrated, M1 macrophages and adipocytes establish a complex reciprocal communication network involving cytokines, chemokines, and microRNA-containing exosomes, among others, that contribute to functional dysregulation [5]. In addition, one key hallmark of adipose tissue inflammation is the presence of histological markers, termed crown-like structures (CLS), generated by proinflammatory macrophages surrounding dead or dying adipocytes [6].
1.3. Obesity-Associated Cancer
The absence of excess body fat lowers the risk for thirteen sites/types of cancer, among which are esophagus adenocarcinoma, colon and rectum, liver, pancreas, breast (postmenopausal), and corpus uteri cancer [7]. Given the wide variety of alterations involved in obesity-related pathophysiology on the one hand and the complexity of the mechanisms that are altered during different carcinogenic processes on the other, it is difficult to address the mechanisms involved in this relationship in a single model. For instance, the development of some types of cancer may be more specifically favored by obesity-related alterations in sex hormone metabolism while others are stimulated by enhanced insulin and insulin growth factor signaling, altered adipokine levels, or elevated proinflammatory and/or oxidative status [8][9].
Interestingly, adipocytes in obesity suffer several subcellular/metabolic alterations, such as mitochondrial dysfunction and endoplasmic reticulum (ER) stress. These changes result in an increased production of reactive oxygen species (ROS) [10][11]. In turn, such constantly elevated ROS production is expected to further damage cell components and threaten cell viability, exacerbating the aforementioned formation of CLS, thereby generating a microenvironment favorable for tumor progression due to its effects on cell proliferation, survival, invasion, and metastasis [12]. For instance, obesity promotes oxidative stress and favors tumor development by inducing DNA damage and genomic instability. Oxidative stress is further increased by mitochondrial alterations and potentiated by obesity-induced chronic inflammation. Moreover, obesity-induced dyslipidemia promotes lipid peroxidation and DNA mutations. Finally, obesity-induced hyperglycemia increases insulin signaling, glycolytic pathway activity, oxidative stress, and DNA alterations (reviewed in [13]).
1.4. Tumor Microenvironment: Adipocytes and Macrophages
As mentioned above, adipose tissue in obesity may undergo several structural, metabolic, and functional changes that create a proinflammatory and oxidative milieu within the tissue microenvironment. These local changes involving cell–cell and cell–extracellular matrix crosstalk are thought to be particularly relevant in tumors that are in close proximity to adipose tissue, as is the case for breast cancer [14].
Multiple players in the obese adipose tissue microenvironment promote insulin resistance and elevate local insulin levels, which then can enhance phosphoinositide 3-kinase (PI3K) signaling and promote tumorigenesis [15]. Moreover, increased vascular endothelial growth factor (VEGF) production associated with adipose extracellular matrix remodeling likely facilitates tumor angiogenesis [16] and tumorigenesis in breast cancer [17]. In addition, factors secreted from adipose tissue that are elevated in obesity, such as leptin, visfatin, or those contained in EVs, have been shown to promote processes associated with tumor development, such as angiogenesis, invasion, viability, metalloproteinase activation, and ROS production, and do so by elevating ERK and PI3K/Akt signaling among other mechanisms [18][19].
Researchers will review the available information relating to endocrine and paracrine communication between adipose tissue and macrophages. The aspects that will be covered in this research are graphically summarized in Figure 1.
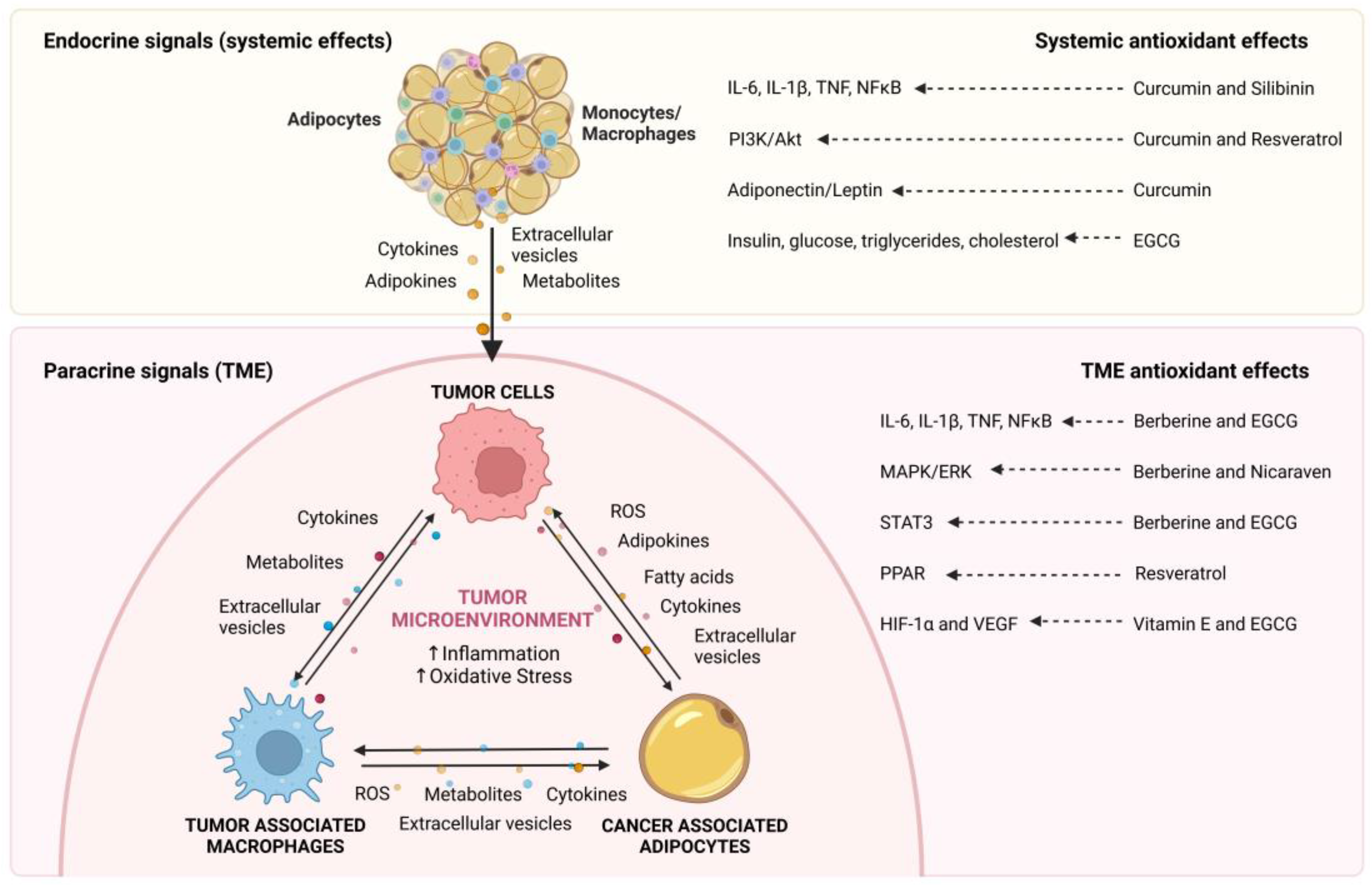
Figure 1. Endocrine and paracrine communication between adipocytes, macrophages, and cancer cells. Obesity-induced changes in the adipose tissue result in increased expression of cytokines, adipokines, metabolites, and extracellular vesicles (EVs) that influence cancer cell behavior and promote tumor development. In addition, in the tumor microenvironment (TME), adipocytes and macrophages are key components that interact with cancer cells by secreting a variety of signaling molecules, including, adipokines, cytokines, metabolites, fatty acids and EVs, which increase local inflammation and oxidative stress to generate a favorable microenvironment for tumor progression.
2. Endocrine Communication between Adipose Tissue and the Tumor Microenvironment
Inflammation-related cytokines, such as interleukin-6 (IL-6) and C-reactive protein (CRP), as well as changes in metabolism-related adipokines, such as adiponectin and leptin, have been shown to reflect a proinflammatory microenvironment that promotes tumor development and progression. Patients with a normal weight tend to have higher levels of the anti-inflammatory and insulin-sensitizing adipokine adiponectin while those who are overweight or obese have higher leptin, IL-6 and CRP levels. In addition, elevated IL-6 is associated with increased cancer mortality in patients with obesity [20]. In the following paragraphs, researchers will focus on the mechanisms connecting adipose tissue to the tumor microenvironment as summarized in Table 1.
Table 1. Endocrine communication between adipose tissue and the tumor microenvironment.
| Cancer Type | Mechanism | Refs. |
|---|---|---|
| Colitis-associated cancer | IL-6, CCL-20/CCR-6 | [21] |
| Colon cancer, colorectal adenocarcinoma, and hepatocellular carcinoma | Survivin | [22] |
| Hepatocellular carcinoma | PI3Kγ, insulinemia and steatosis | [23] |
| Liver and uterine cancer | microRNA, fatty acid and arachidonic acid metabolism, cell growth, and inflammation | [24] |
| Endometrial Cancer | Leptin, proinflammatory cytokines (IL-6) and oxidative stress | [25] |
| Breast cancer | Leptin | [26] |
| Breast cancer | NLRC4, IL-1β, VEGF, angiogenesis | [27] |
| Breast cancer | ANGPTL4, IL-1β, angiogenesis, macrophage tumor infiltration | [28] |
| Breast cancer | PPARα and FAK-signaling (inflammation and lipid metabolism, such as PPARα, IL-1β, and ANGPTL4) | [29] |
| Breast cancer | White adipose tissue inflammation, higher serum lipid, glucose, C-reactive protein levels and CLS | [30] |
| Breast cancer | White adipose tissue inflammation, CLS, insulin, glucose, triglycerides, HDL, leptin, adiponectin, C-reactive protein, and IL-6 | [31] |
| Breast cancer | GM-CSF and MMP-9 | [32] |
| Breast cancer | IL-5 and GM-CSF | [33] |
| Lung cancer | Lipid metabolism and oxidative stress | [34] |
| Prostate Cancer | IL-6, MDSCs, M2/M1 tumor infiltration, pSTAT3 | [35] |
| Prostate Cancer | IL-6, VEGF, CXCL1/2/13 | [36] |
Overweight and obese breast cancer patients develop CLS+ and adipose tissue inflammation, which is associated with markers of metabolic derangements, such as increased lipid, glucose and CRP levels in serum [37]. Moreover, breast cancer patients with adipose inflammation have increased insulin, glucose, leptin, triglycerides, CRP, and IL-6 levels as well as decreased HDL and adiponectin levels. Furthermore, obesity, metabolic syndrome and the circulation of proinflammatory molecules are associated with poorer prognosis in breast cancer patients [31].
In lung cancer, there is also a strong association between overweight/obesity, lipid alterations, and oxidative stress. Indeed, the total cholesterol/HDL ratio, triglyceride levels, lipid peroxidation and oxidative stress are increased in lung cancer patients compared to healthy people. Moreover, these alterations increase in lung cancer patients who are overweight and obese. These data suggest that obesity could enhance lung cancer-induced systemic changes, such as lipid and redox imbalance [37].
A high-fat diet (HFD) enhances the M2/M1 macrophage ratio and accelerates prostate cancer tumor growth via IL-6/STAT3 signaling [37]. Furthermore, HFD-induced obese mice showed increases in VEGF, IL-6, chemokine ligand 2 (CCL2), and C-X-C motif chemokine ligand 1/2/13 (CXCL1/2/13), which promote angiogenesis and the infiltration of immune cells that favor tumor development and metastasis [36].
Interestingly, in endometrial cancer patients, body mass index (BMI), leptin, IL-6 and oxidative stress correlate with pathogenesis and poor prognosis [25]. Likewise, in breast cancer, several factors secreted by adipose tissue augment cancer cell aggressiveness. For instance, leptin secreted by adipose stromal/stem cells isolated from women with obesity enhances the expression of epithelial-to-mesenchymal transition (EMT) and metastasis-related genes, such as serpine 1, matrix metalloproteinase 2 (MMP-2) and IL-6 [26]. In addition, the reciprocal relationship between adipocytes, immune cells and cancer cells induces a systemically immunosuppressive macroenvironment that promotes breast cancer development. Moreover, breast cancer cells implanted in a mouse model of diet-induced obesity modified the visceral adipose tissue, spleen and tumor microenvironment in order to create an immunosuppressive milieu that promoted cancer development. The characteristic increase in the M1/M2 macrophage ratio reported in the visceral adipose tissue in obesity was completely reversed in tumor-bearing mice, resulting in a predominantly M2-polarized profile typically observed in the tumor microenvironment. Altogether, these results suggest the existence of a regulatory feedback mechanism between the growing tumor and adipose tissue, which generates local and systemic immunosuppressive conditions that promote tumor development [37].
In addition, the obese tumor microenvironment recruits macrophages that activate the NOD-like receptor 4 (NLRC4) inflammasome, leading to interleukin-1β (IL-1β) activation. IL-1β promotes breast cancer progression by increasing adipocyte VEGF secretion and angiogenesis [27]. Moreover, angiopoietin-like 4 (ANGPTL4), a known proangiogenic factor in cancer, is induced by IL-1β from adipocytes in a manner dependent on NF-κB and MAPK. In this way, adipocyte-secreted ANGPTL4 promoted angiogenesis and breast cancer progression in obese mice [37]. Furthermore, factors secreted by adipose tissue from HFD-induced obese mice and the fat tissue of female patients with obesity led to the upregulation of genes involved in inflammation and lipid metabolism, such as PPARα, IL-1β and ANGPTL4 in triple-negative breast cancer cells. Moreover, focal adhesion kinase (FAK) activation increased in these cancer cells, resulting in elevated lipid metabolism, wound healing, proliferation, and invasion [29].
Regarding the role of specific proinflammatory cytokines and adipokines, a study in a mouse model of chemically induced colitis-associated colorectal cancer (CAC) showed that diet-induced obesity increased IL-6 levels, thereby favoring macrophage polarization towards tumor-promoting M2 macrophages. Moreover, M2 macrophages produced CC-chemokine-ligand-20 (CCL-20) that promoted the recruitment of CC-chemokine-receptor-6 (CCR-6)-expressing B and T cells. The recruited B cells promoted further macrophage polarization and the T cells inhibited other immune cells, thereby favoring CAC progression [21]. Survivin is also secreted by adipose tissue and drives protumoral macrophage polarization in colon cancer, colorectal adenocarcinoma and hepatocellular carcinoma. Indeed, adipose-derived stem cells (ASCs) from subjects with obesity release survivin that induces tumor-associated macrophage (TAM) reprogramming coincident with increased survivin expression. Thus, survivin produced by ASCs and macrophages together promotes the malignancy of colon and liver cancer cells [22].
3. Paracrine Communication between Adipocytes, Macrophages and Cancer Cells in the Tumor Microenvironment
Tumors contain many different cell populations beyond cancer cells, including fibroblasts, immune cells (macrophages, lymphocytes, neutrophils), endothelial cells and adipocytes, among others [38][39]. Moreover, these additional populations of tumor stromal cells promote extracellular matrix remodeling, cell migration, angiogenesis, invasion, metastasis, and drug resistance by producing a variety of signaling molecules that include extracellular matrix constituents, growth factors, metabolites, chemokines and cytokines [40]. Therefore, to determine what drives tumor development, it is necessary to understand how the complex communication between tumor cells and the surrounding stroma components conspires to produce factors that augment inflammation in the tumor microenvironment (TME), thereby favoring tumor progression and metastasis. Note that articles reporting on the paracrine communication within the tumor microenvironment are summarized in Table 2.
Table 2. Paracrine communication between adipocytes, macrophages, and cancer cells in the tumor microenvironment.
| Signals from Adipocytes to Cancer Cells | |||
| Cancer Type | Mechanisms | Effect on TME | Refs. |
| Breast cancer | β-hydroxybutyrate increases histone acetylation and upregulates the expression of tumor-promoting genes (IL-1β and LCN2) | Tumor growth and poor prognosis | [41] |
| Breast cancer | A positive feedback loop between the cytokines LIF and CXCLs | Invasion and metastasis | [42] |
| Breast cancer | G-CSF/Stat3 | EMT, migration, and invasion | [43] |
| Breast cancer | Upregulation of MMP9, TWIST1 and Vimentin | EMT, migration, and invasion | [44] |
| Breast cancer | IGFBP-2 and MMP-2 | Adipocytes acquire CAA-like phenotype. Migration, invasion, and metastasis. | [45] |
| Breast cancer | IL-8 | Angiogenesis and breast cancer cell dissemination | [46] |
| Breast cancer | IL-8/STAT3 | Angiogenesis and tumorigenesis | [47] |
| Breast cancer | Cytokines (CCL2, CCL5, IL-6, IL-8, CXCL10) activate Src to upregulate Sox2 and induce miR-302b | Cancer stem cell self-renewal and metastasis | [48] |
| Breast cancer | Increase expression of MMP-11, IL-6, and IL-1β | Invasion and metastasis | [49] |
| Breast cancer | ATGL/HSL-mediated fatty acids released by adipocytes. FAO induction through increased CPT1A in cancer cells. |
Proliferation and migration | [50] |
| Breast cancer | ATGL-dependent lipolysis and uncoupled FAO | Invasion | [51] |
| Ovarian cancer | FABP4 | Metastasis and Carboplatin resistance | [52] |
| Ovarian cancer | Exosomal miR21/APAF1 | Invasion and Paclitaxel resistance | [53] |
| Ovarian cancer | CD36/Exogenous fatty acid uptake | Migration, invasion, and metastasis | [54] |
| Ovarian cancer | Arachidonic acid/Akt pathway | Cisplatin resistance | [55] |
| Melanoma | Adipocyte-EVs transfer proteins implicated in FAO | Metabolic reprogramming, migration and invasion | [56] |
| Melanoma | Expression of EMT genes (SNAI1, MMP-9, TWIST and Vimentin) | Invasion | [57] |
| Melanoma | PI3K/Akt and MEK/ERK pathways | Resistance to chemotherapeutic drugs (Cisplatin and Docetaxel) | [58] |
| Prostate cancer | CCR3/CCL7 axis | Migration | [59] |
| Gastric cancer | Oleic acid/PI3K/Akt/MMP2 | Increase in lipid uptake and invasion | [60] |
| Gastric cancer | DGAT2-dependent lipid droplet accumulation and redox homeostasis | Proliferation, migration and invasion | [61] |
| Colon cancer | Upregulation of autophagy and mitochondrial FAO via AMPK | Migration and EMT | [62] |
| Colon cancer | CPT1A-dependent FAO promote the acetylation and nuclear translocation of β-catenin | Promote cancer stem cell functions, cell proliferation and tumor growth | [63] |
| Pancreatic cancer | SAA1 | Adipocytes acquire CAA-like phenotype and induce migration, invasion, and EMT in cancer cells | [64] |
| Oral squamous cell carcinoma, melanoma, breast cancer | CD36/FAO-related genes | Poor prognosis. Lymph node metastasis | [65] |
| Signals from Macrophages to Cancer Cells | |||
| Cancer Type | Mechanisms | Effect on TME | Refs. |
| Breast cancer | WNT7B /VEGFA | Angiogenesis, tumor growth, invasion and metastasis | [66] |
| Breast cancer | Lactate and CCL5-CCR5 axis | Aerobic glycolysis and EMT | [67] |
| Pancreatic, breast, and lung cancer | IL-4-inducing cathepsin activity in macrophages | Tumor growth, angiogenesis and invasion | [68] |
| Pancreatic ductal adenocarcinoma | M2-polarized TAMs, angiopoietin-axis and TIE2-expressing monocytes | Angiogenesis and metastasis | [69] |
| Lung cancer | AMPK/PGC-1α, TNFα, PD-L1 | Aerobic glycolysis, tumor hypoxia and resistance to anticancer therapies | [70] |
AMPK: AMP-activated protein kinase; ANGPTL4: angiopoietin-like 4; APAF1: apoptotic peptidase activator factor 1; ATGL: adipose triglyceride lipase; CCL2: CC-chemokine-ligand-2; CCL5: CC-chemokine-ligand-5; CCR3: CC-chemokine-receptor-3; CCR5: CC-chemokine-receptor-5; Chk1: checkpoint kinase 1; CPT1A: carnitine palmitoyl transferase I; CXCLs: C-X-C motif chemokine ligands; DGAT2: diacylglycerol acyltransferase 2; EMT: epithelial-to-mesenchymal transition; EVs: extracellular vesicles; G-CSF: granulocyte colony-stimulating factor; IGFBP-2: insulin-like growth factor binding protein 2; IL-1β: interleukin-1β; IL-6: interleukin-6; IL-8: interleukin-8; LCN2: lipocalin 2; LIF: leukemia inhibitory factor; MMPs: matrix metalloproteinase; NLRC4: NOD-like receptor 4; PD-1: programmed cell death-1; PD-L1: programmed death ligand 1; PGC-1α: peroxisome proliferator-activated coactivator gamma 1-alpha; PPARγ: peroxisome proliferator-activated receptor gamma; SAA1: serum amyloid A1; TNFα: tumor necrosis factor alpha; TWIST1: twist-related protein 1; VEGFA: vascular endothelial growth factor A.
3.1. Effects of Adipocytes on Cancer Cells
Adipocytes play a fundamental role in the TME of multiple types of cancer. Available evidence indicates that dynamic communication between tumor cells and adipocytes leads to functional changes in both cell types that can drive tumor progression. Adipocytes have been shown to produce factors involved in matrix remodeling, metabolic reprogramming, EMT, angiogenesis, invasion and the survival of cancer cells as well as drug resistance [53][59][62][71].
Cancer-associated adipocytes (CAAs) produce elevated insulin-like growth factor binding protein 2 (IGFBP-2) levels in the tumor microenvironment, which upregulates MMP-2 and then promotes migration, invasion, and metastasis in breast cancer [45]. Likewise, CAAs cocultured with breast cancer cells have been shown to increase their expression of MMP-11 as well as proinflammatory cytokines (IL-6 and IL-1β) and thereby promote tumor cell invasion and metastasis [49]. Other investigators demonstrated that cancer cells cultured with in vitro differentiated adipocytes or proinflammatory cytokines (IL-6, IL-8, CXCL10, CCL2 or CCL5) activate Src to upregulate Sox2, generating feed-forward loops that contribute to mammary cancer stem cell self-renewal and drive metastatic tumor progression [48]. Moreover, the interaction between CAAs and breast cancer cells involves a positive feedback loop between the cytokine leukemia inhibitory factor (LIF) and CXCL subfamily chemokines, which promotes breast cancer invasion and metastasis [42].
The evidence also suggests that adipocytes mediate proangiogenic events in several tumors. CAAs secrete high levels of IL-8, which increases angiogenesis at the primary tumor site, induces a protumoral neutrophil phenotype, modifies the expression of cell-adhesion molecules in tumor cells and favors early dissemination of breast cancer [46]. Furthermore, Al-Khalaf et al. reported that CAAs secreted high levels of IL-8, which was crucial for enhancing the proangiogenic effects of breast adipocytes [47].
Interestingly, adipocytes induce metabolic reprogramming in tumor cells. Peritoneum-derived adipocytes induce lipid droplet accumulation and fatty acid oxidation (FAO) in gastric cancer cells through the upregulation of diacylglycerol acyltransferase 2 (DGAT2) in a CCAAT/enhancer-binding protein alpha (C/EBPα)-dependent manner. The lipid-rich environment stimulates nicotinamide adenine dinucleotide phosphate (NADPH) production, promotes resistance to anoikis through ROS-dependent mechanisms, and supports HFD-induced peritoneal dissemination and lung metastasis in vivo [61]. Mammary adipocytes stimulate metabolic remodeling in cancer cells. Indeed, fatty acid metabolism in cancer cells is uncoupled from ATP production, leading to the promotion of the Warburg effect and increased glycolysis, thereby promoting the proliferation, migration and invasion of cancer cells [51]. Furthermore, human omental adipocytes may act as a source of oleic acid for gastric cancer cells in omental gastric cancer metastases. Oleic acid enhances the invasiveness of gastric cancer cells, as well as the expression of MMP-2, by activating the PI3K-Akt signaling pathway in a PTEN-independent manner [60]. Primary mammary gland-derived adipocytes promote the tumorigenicity of cancer cells that express monocarboxylate transporter 2 (MCT2) via β-hydroxybutyrate. In detail, β-hydroxybutyrate secreted from adipocytes increases histone H3K9 acetylation and induces IL-1β and lipocalin 2 (LCN2) expression to enhance tumorigenicity in MCT2-expressing breast cancer cells, which in turn is linked to poor prognosis in breast cancer patients [41].
The fatty acid receptor CD36 is overexpressed in many metastatic tumors, such as oral squamous cell carcinomas (OSCC), melanoma and breast cancers. In OSCC cell lines or patient-derived cells with low metastatic potential, high levels of CD36 significantly increased lymph node metastasis. Furthermore, metastatic potential depends on the increased expression of FAO-related genes and is boosted by an HFD in a CD36-dependent manner. Consistent with these data, CD36 expression strongly correlates with poor prognosis in lung, bladder and breast cancer patients [65]. Later, the same group observed that ovarian cancer cells cocultured with primary human omental adipocytes expressed high levels of CD36, thereby facilitating exogenous fatty acid uptake and ovarian cancer metastasis. Conversely, CD36 inhibition reduced microenvironment-derived fatty acid uptake in ovarian cancer cells, decreasing adipocyte-stimulated invasion and migration in vitro, as well as tumor growth in vivo [54]. These observations highlight the crucial role of lipid metabolism in tumor progression and metastasis.
Additionally, adipocytes can induce EMT in several types of cancer. In breast cancer cells, mature adipocytes induce the EMT phenotype and promote cancer cell migration, invasion and proliferation, which could be associated with the upregulation of MMP-9 and twist-related protein 1 (TWIST1) [44]. Melanoma cells cocultured with differentiated 3T3-L1 adipocyte cells increased their invasive ability by promoting the expression of EMT genes (SNAI1, MMP-9, TWIST, and vimentin) and decreased the expression of genes, such as E-cadherin and Kiss1 [57]. Similarly, the conditioned medium of CAAs has been shown to improve migration/invasion and chemoresistance, as well as to promote EMT in pancreatic cancer cells by the upregulation of serum amyloid A1 (SAA1) [64], which has been associated with poor prognosis in breast cancer [72].
Other studies related to therapy resistance established that the conditioned media from human adipocytes promote resistance of melanoma cells to chemotherapeutic drugs (cisplatin and docetaxel) and therapeutic agents targeting the PI3K/Akt and MEK/ERK pathways [58]. Similarly, another study showed that human adipocytes of both subcutaneous and visceral/omental origin secrete soluble factors that increase resistance to chemotherapeutic drugs in ovarian cancer cells by activating the Akt pathway [55]. Moreover, primary human omental adipocytes induce FABP4 expression in ovarian cancer cells and promote metastasis and Carboplatin resistance in ovarian cancer cells [52]. Together, these studies highlight the role of adipocytes in inducing responses in tumor cells that enhance therapy resistance.
Finally, recent studies showed that murine and human adipocytes release EVs that deliver enzymes and substrates (fatty acids) involved in FAO to melanoma cells, which subsequently improves mitochondrial metabolism and remodels the mitochondrial network to support melanoma migration and invasion [73]. Another study reported that EVs from CAAs delivered microRNA-21 (miR21) to ovarian cancer cells, where it suppresses apoptosis and induces chemoresistance to paclitaxel through the downregulation of miR21’s direct target apoptotic peptidase activator factor 1 (APAF1) [53].
3.2. Effects of Tumor-Associated Macrophages on Cancer Cells
As mentioned above, the communication between cancer cells and TAMs is bidirectional. In the TME, TAMs typically promote cancer cell proliferation, angiogenesis, metastasis, and other processes through various anti-inflammatory mechanisms [74].
TAMs are known to be strongly involved in tumor angiogenesis. For instance, it has been reported that the presence of M2-polarized TAMs and Tie2-expressing monocytes is associated with angiogenesis, as well as decreased overall and recurrence-free survival in human PDAC patients [69]. In pancreatic islet cancers, breast tumors and lung metastases, interleukin-4 (IL-4) induces cathepsin protease activity in macrophages. Consequently, cathepsins B and S supplied by TAMs promote tumor growth, angiogenesis and invasion in vivo, as well as markedly increase the invasiveness of cancer cells in vitro [68]. Others reported that the development of human breast cancer is strongly associated with overexpression of the Wnt family ligand Wnt7b in TAMs in human breast carcinoma. Furthermore, in mice, Wnt7b was shown to play a crucial role in the malignant progression of luminal breast cancer by promoting angiogenesis, tumor growth, progression, invasion and metastasis [66].
M2-like TAMs reportedly promote glycolysis and hypoxia in cancer cells, leading to changes in cellular metabolism that are necessary for tumor initiation and progression. In non-small cell lung cancer, TAMs secrete TNFα to promote glycolysis in tumor cells and activate both AMPK and peroxisome proliferator-activated coactivator gamma 1-alpha, which together augment tumor hypoxia by promoting mitochondrial oxygen consumption and increasing mitochondrial membrane potential [70]. Furthermore, in this same study, TAMs were found to significantly reduce T cell infiltration by upregulating programmed death ligand 1 (PD-L1) expression in tumors. Therefore, the immune mechanisms that are associated with PD-L1 expression and controlled by TAMs can be considered critical for tumor immune escape [75]. Importantly, lactate efflux plays a critical role in tumor–stroma communication. Cancer cell-derived lactate increases the secretion of CCL5 through Notch signaling in TAMs, and CCL5 in turn induces EMT and aerobic glycolysis in breast cancer cells. In addition, TGF-β signaling regulates the expression of the CCL5-CCR5 axis components, which induce aerobic glycolysis in cancer cells by activating AMPK signaling. These observations point towards a pivotal role for the CCL5-CCR5 axis in metabolic communication between cancer cells and macrophages [67].
Finally, differences between TAMs from primary and metastatic cancers have been reported. In the primary tumor, TNFα induces EMT and the expression of IL12Rβ, a subunit of the IL-35 receptor, to promote tumor cell migration and invasion. Instead, at metastatic sites, TAMs secrete IL-35 to activate the JAK2–STAT6–GATA3 signaling axis in cancer cells, which reverses EMT to facilitate tissue colonization by cancer cells [76]. Additionally, Morrissey et al. reported that tumor-derived EVs polarize premetastatic niche macrophages towards an immunosuppressive phenotype through NF-kB-dependent glycolytic reprogramming to favor tumor cell metastasis. These prometastatic macrophages are characterized by increased de novo synthesis of PD-L1, increased glucose uptake, and GLUT-1 expression as well as the increased conversion of pyruvate to lactate, which subsequently augments further PD-L1 expression [77].
This entry is adapted from the peer-reviewed paper 10.3390/antiox12010126
References
- Nicholas S Hendren; Association of Body Mass Index and Age With Morbidity and Mortality in Patients Hospitalized With COVID-19: Results From the American Heart Association COVID-19 Cardiovascular Disease Registry. Circulation 2021, 143, 135-144, 10.1161/CIRCULATIONAHA.120.051936.
- G S Hotamisligi; Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993, 5091, 87-91, 10.1126/science.7678183.
- Jan-Bernd Funcke; Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication. J Lipid Res 2019, 10, 1648-1684, 10.1194/jlr.R094060.
- Aaron R Cox; Immune Cells Gate White Adipose Tissue Expansion. Endocrinology 2019, 160, 1645-1658, 10.1210/en.2019-00266.
- Zhaohua Cai; New Insights into Adipose Tissue Macrophages in Obesity and Insulin Resistance. Cells 2022, 11, 1424, 10.3390/cells11091424.
- Kim-Anne Lê; Subcutaneous Adipose Tissue Macrophage Infiltration Is Associated With Hepatic and Visceral Fat Deposition, Hyperinsulinemia, and Stimulation of NF-κB Stress Pathway. Diabetes 2011, 60, 2802–2809, https://doi.org/10.2337/db10-1263.
- Béatrice Lauby-Secretan; Body Fatness and Cancer — Viewpoint of the IARC Working Group. N Engl J Med 2016, 375, 794-798, 10.1056/NEJMsr1606602.
- Carolin Maria Frisch; Non-small cell lung cancer cell survival crucially depends on functional insulin receptors. Endocrine-Related Cancer 2015, 22, 609–621, https://doi.org/10.1530/ERC-14-0581.
- Aneta Słabuszewska-Jóźwiak; Role of Leptin and Adiponectin in Endometrial Cancer. Int. J. Mol. Sci 2022, 23, 5307, https://doi.org/10.3390/ijms23105307.
- Amy K.Hauck; Adipose oxidative stress and protein carbonylation. JBC Reviews 2019, 294, 1083-1088, https://doi.org/10.1074/jbc.R118.003214.
- Laura Jackisch; Tunicamycin-Induced Endoplasmic Reticulum Stress Mediates Mitochondrial Dysfunction in Human Adipocytes. The Journal of Clinical Endocrinology & Metabolism 2020, 105, 2905–2918, https://doi.org/10.1210/clinem/dgaa258.
- MakiyaNishikawa; Catalase delivery for inhibiting ROS-mediated tissue injury and tumor metastasis. Advanced Drug Delivery Reviews 2009, 61, 319-326, https://doi.org/10.1016/j.addr.2009.01.001.
- MoonisahUsman; DNA damage in obesity: Initiator, promoter and predictor of cancer. Mutation Research/Reviews in Mutation Research 2018, 778, 23-37, https://doi.org/10.1016/j.mrrev.2018.08.002.
- Quail, D.F; The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol 2019, 15, 139–154, https://doi.org/10.1038/s41574-018-0126-x.
- Yu Lu; Insulin receptor tyrosine kinase substrate (IRTKS) promotes the tumorigenesis of pancreatic cancer via PI3K/AKT signaling. Human Cell volume 2022, 35, 1885–1899, https://doi.org/10.1007/s13577-022-00770-w.
- Jian-Wei Gu; Postmenopausal obesity promotes tumor angiogenesis and breast cancer progression in mice. Cancer Biology & Therapy 2011, 15, 398-408, https://doi.org/10.4161/cbt.11.10.15473.
- BO RI SEO; Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. SCIENCE TRANSLATIONAL MEDICINE 2015, 7, 301, 10.1126/scitranslmed.3010467.
- Ruben Rene Gonzalez-Perez; Leptin’s Pro-Angiogenic Signature in Breast Cancer. Cancers 2013, 5, 1140-1162, https://doi.org/10.3390/cancers5031140.
- Isadora Ramos-Andrade; Obese adipose tissue extracellular vesicles raise breast cancer cell malignancy. Endocrine-Related Cancer 2020, 27, 571–582, Endocrine-Related Cancer.
- Daniel T Dibaba; Association between obesity and biomarkers of inflammation and metabolism with cancer mortality in a prospective cohort study. Metabolism 2019, 94, 69-76, 10.1016/j.metabol.2019.01.007.
- Claudia M Wunderlich; Obesity exacerbates colitis-associated cancer via IL-6-regulated macrophage polarisation and CCL-20/CCR-6-mediated lymphocyte recruitment. Nat Commun 2018, 9, 1646, 10.1038/s41467-018-03773-0.
- Benaiges, E.; Survivin drives tumor-associated macrophage reprogramming: a novel mechanism with potential impact for obesity. Cell Oncol 2021, 44, 777–792, https://doi.org/10.1007/s13402-021-00597-x.
- Barbara Becattini; PI3Kγ promotes obesity-associated hepatocellular carcinoma by regulating metabolism and inflammation. JHEP Rep 2021, 3, 100359, 10.1016/j.jhepr.2021.100359.
- Haluk Dogan; Elucidation of molecular links between obesity and cancer through microRNA regulation. BMC Med Genomics 2020, 13, 161, 10.1186/s12920-020-00797-8.
- Clelia Madeddu; Correlation of Leptin, Proinflammatory Cytokines and Oxidative Stress with Tumor Size and Disease Stage of Endometrioid (Type I) Endometrial Cancer and Review of the Underlying Mechanisms. Cancers (Basel) 2022, 14, 268, 10.3390/cancers14020268.
- Strong, A.L; Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Res 2015, 17, 112, https://doi.org/10.1186/s13058-015-0622-z.
- Kolb, R; Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun 2016, 7, 13007, https://doi.org/10.1038/ncomms13007.
- Kolb, R; Obesity-associated inflammation promotes angiogenesis and breast cancer via angiopoietin-like 4. Oncogene 2019, 38, 2351–2363, https://doi.org/10.1038/s41388-018-0592-6.
- Christina Blücher; Secreted Factors from Adipose Tissue Reprogram Tumor Lipid Metabolism and Induce Motility by Modulating PPARα/ANGPTL4 and FAK. Mol Cancer Res 2020, 18, 1849-1862, 10.1158/1541-7786.MCR-19-1223.
- Yi-Xin Zhao; The Relationship Between White Adipose Tissue Inflammation and Overweight/Obesity in Chinese Female Breast Cancer: A Retrospective Study. Adv Ther 2020, 37, 2734-2747, 10.1007/s12325-020-01368-0.
- Neil M Iyengar; Systemic Correlates of White Adipose Tissue Inflammation in Early-Stage Breast Cancer. Clin Cancer Res 2016, 22, 2283-9, 10.1158/1078-0432.CCR-15-2239.
- Francesca Reggiani; Adipose Progenitor Cell Secretion of GM-CSF and MMP9 Promotes a Stromal and Immunological Microenvironment That Supports Breast Cancer Progression. Cancer Res 2017, 77, 5169-5182, https://doi.org/10.1158/0008-5472.CAN-17-0914.
- Daniela F Quail; Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol 2017, 19, 974-987, 10.1038/ncb3578.
- Katarzyna Zabłocka-Słowińska; Oxidative stress in lung cancer patients is associated with altered serum markers of lipid metabolism. PLoS ONE 2019, 14, e0215246, https://doi.org/10.1371/journal.pone.0215246.
- Takuji Hayashi; High-Fat Diet-Induced Inflammation Accelerates Prostate Cancer Growth via IL6 Signaling. Clin Cancer Res 2018, 24, 4309–4318, https://doi.org/10.1158/1078-0432.CCR-18-0106.
- Han Jin Cho; A High-Fat Diet Containing Lard Accelerates Prostate Cancer Progression and Reduces Survival Rate in Mice: Possible Contribution of Adipose Tissue-Derived Cytokines. Nutrients 2015, 7, 2539-2561, https://doi.org/10.3390/nu7042539.
- Aleida Núñez-Ruiz; Obesity modulates the immune macroenvironment associated with breast cancer development. PLoS ONE 2022, 17, e0266827, https://doi.org/10.1371/journal.pone.0266827.
- Quail D.F.; Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437, 10.1038/nm.3394.
- Hanahan D.; Hallmarks of cancer: The next generation.. Cell 2011, 144, 646–674, 10.1016/j.cell.2011.02.013.
- Hanahan D.; Accessories to the crime: Functions of cells recruited to the tumor microenvironment.. Cancer Cell 2012, 21, 309–322, 10.1016/j.ccr.2012.02.022.
- Huang C.K.; Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via beta-hydroxybutyrate. Nat. Commun. 2017, 8, 14706, 10.1038/ncomms14706.
- Zhou C.; Cancer-associated adipocytes promote the invasion and metastasis in breast cancer through LIF/CXCLs positive feedback loop. Int. J. Biol. Sci 2022, 18, 1363–1380, 10.7150/ijbs.65227.
- Liu L.; Cancer-associated adipocyte-derived G-CSF promotes breast cancer malignancy via Stat3 signaling. J. Mol. Cell. Biol. 2020, 12, 723–737, 10.1093/jmcb/mjaa016.
- Lee Y.; Adipocytes can induce epithelial-mesenchymal transition in breast cancer cells. Breast Cancer Res. Treat. 2015, 153, 323–335, 10.1007/s10549-015-3550-9.
- Wang C.; Human adipocytes stimulate invasion of breast cancer MCF-7 cells by secreting IGFBP-2. PLoS ONE 2015, 10, e0119348, 10.1371/journal.pone.0119348.
- Vazquez Rodriguez G.; Adipocytes Promote Early Steps of Breast Cancer Cell Dissemination via Interleukin-8. Front. Immunol. 2018, 9, 1767, 10.3389/fimmu.2018.01767.
- Al-Khalaf H.H.; Interleukin-8 Activates Breast Cancer-Associated Adipocytes and Promotes Their Angiogenesis- and Tumorigenesis-Promoting Effects. Mol. Cell Biol. 2019, 39, e00332-18, 10.1128/MCB.00332-18.
- Picon-Ruiz M.; Interactions between Adipocytes and Breast Cancer Cells Stimulate Cytokine Production and Drive Src/Sox2/miR-302b-Mediated Malignant Progression. Cancer Res. 2016, 76, 491–504, 10.1158/0008-5472.CAN-15-0927.
- Dirat B.; Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465, 10.1158/0008-5472.CAN-10-3323.
- Balaban S.; Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1, 10.1186/s40170-016-0163-7.
- Wang Y.Y.; Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight. 2017, 2, e87489, 10.1172/jci.insight.87489.
- Mukherjee A.; Adipocyte-Induced FABP4 Expression in Ovarian Cancer Cells Promotes Metastasis and Mediates Carboplatin Resistance. Cancer Res. 2020, 80, 1748–1761, 10.1158/0008-5472.CAN-19-1999.
- Au Yeung C.L.; Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150, 10.1038/ncomms11150.
- Ladanyi A.; Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301, 10.1038/s41388-017-0093-z.
- Yang J.; Adipocytes promote ovarian cancer chemoresistance. Sci. Rep. 2019, 9, 13316, 10.1038/s41598-019-49649-1.
- Lazar I.; dipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation: A Novel Mechanism Linking Obesity and Cancer. Cancer Res. 2016, 76, 4051–4057, 10.1158/0008-5472.CAN-16-0651.
- Kushiro K.; Adipocytes Promote B16BL6 Melanoma Cell Invasion and the Epithelial-to-Mesenchymal Transition. Kushiro K. 2012, 5, 73-82, 10.1007/s12307-011-0087-2.
- Chi M.; Adipocytes contribute to resistance of human melanoma cells to chemotherapy and targeted therapy. Curr. Med. Chem. 2014, 21, 1255–1267, 10.2174/0929867321666131129114742.
- Laurent V.; Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230, 10.1038/ncomms10230.
- Xiang F.; Omental adipocytes enhance the invasiveness of gastric cancer cells by oleic acid-induced activation of the PI3K-Akt signaling pathway. Int. J. Biochem. Cell. Biol. 2017, 84, 14–21, 10.1016/j.biocel.2016.12.002.
- Li S.; Obesity promotes gastric cancer metastasis via diacylglycerol acyltransferase 2-dependent lipid droplets accumulation and redox homeostasis. Redox Biol. 2020, 36, 101596, 10.1016/j.redox.2020.101596.
- Wen Y.A.; Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593, 10.1038/cddis.2017.21.
- Xiong X.; Upregulation of CPT1A is essential for the tumor-promoting effect of adipocytes in colon cancer. Cell Death Dis. 2020, 11, 736, 10.1038/s41419-020-02936-6.
- Takehara M.; Cancer-associated adipocytes promote pancreatic cancer progression through SAA1 expression. Cancer Sci. 2020, 111, 2883–2894, 10.1111/cas.14527.
- Pascual G.; Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41-45, 10.1038/nature20791.
- Yeo E.J.; Myeloid WNT7b mediates the angiogenic switch and metastasis in breast cancer. Cancer Res. 2014, 74, 2962–2973, 10.1158/0008-5472.CAN-13-2421.
- Lin S.; Lactate-activated macrophages induced aerobic glycolysis and epithelial-mesenchymal transition in breast cancer by regulation of CCL5-CCR5 axis: A positive metabolic feedback loop. Oncotarget 2017, 8, 110426–110443, 10.18632/oncotarget.22786.
- Gocheva V.; IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241-255, 10.1101/gad.1874010.
- Atanasov G.; TIE2-expressing monocytes and M2-polarized macrophages impact survival and correlate with angiogenesis in adenocarcinoma of the pancreas. Oncotarget 2018, 9, 29715–29726, 10.18632/oncotarget.25690.
- Jeong H.; Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res. 2019, 79, 795–806, 10.1158/0008-5472.CAN-18-2545.
- Arendt L.M.; Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093, 10.1158/0008-5472.CAN-13-0926.
- Yang M.; Serum amyloid A expression in the breast cancer tissue is associated with poor prognosis. Oncotarget 2016, 7, 35843–35852, 10.18632/oncotarget.856.
- Clement E.; Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525, 10.15252/embj.2019102525.
- Mantovani A.; Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549-555, 10.1016/S1471-4906(02)02302-5.
- Topalian S.L.; Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012, 24, 207-212, 10.1016/j.coi.2011.12.009.
- Lee C.C.; Macrophage-secreted interleukin-35 regulates cancer cell plasticity to facilitate metastatic colonization. Nat. Commun. 2018, 9, 3763, 10.1038/s41467-018-06268-0.
- Morrissey S.M.; Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 2021, 33, 2040–2058 e2010, 10.1016/j.cmet.2021.09.002.
This entry is offline, you can click here to edit this entry!