Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Succinate dehydrogenase (SDH) is one of the enzymes of the tricarboxylic acid cycle (Krebs cycle) and complex II of the mitochondrial respiratory chain. Succinate dehydrogenase by pesticides (SDHIs) constitute a class of pesticides to fight against fungi. This represents roughly a dozen different molecules sharing the property to inhibit the succinate dehydrogenase (SDH), an enzyme implicated in carbon metabolism and cellular respiration.
- mitochondria
- tricarboxylic acid cycle
- metabolic syndrome
1. Introduction
Succinate dehydrogenase by pesticides (SDHIs) constitute a class of pesticides to fight against fungi. This represents roughly a dozen different molecules sharing the property to inhibit the succinate dehydrogenase (SDH), an enzyme implicated in carbon metabolism and cellular respiration. SDH is also known as complex II of the mitochondrial respiratory chain (MRC). Actually, SDH oxidizes the succinate into fumarate to feed mitochondrial respiration. Consequently, the inhibition of SDH is expected to lower cellular consumption of succinate (that may accumulate) and to impair cellular respiration. Inhibition of SDH is most likely to ground the fungicide effect of SDHIs, and acquired resistance to SDHI in pests correlates with mutations in SDH genes [1]. Experimental evidences in vitro indicate, from the earliest reports with carboxin [2], that SDHI inhibited the SDH of mammals. This has been reexamined recently with eight SDHIs actually in use [3]. This recent report boosted the controversy about the use of these pesticides because it could be considered that extended use of a toxicant to human mitochondrial oxidative metabolism would inevitably result in deleterious consequences in the long term, and/or in more sensitive individuals. These alterations could be anticipated from existing knowledge about pathologies resulting from genetic inactivation of SDH, and from the scientific literature discussing other inhibitors of SDH. At this point it should be highlighted that few mechanistic studies on the impact of different SDHI pesticides on SDH and mitochondrial respiration are presently available. With regard to pesticides acting through inhibition of mitochondrial respiration and the apparent resistance of human beings in contrast with target species, a past example is provided by the prolonged use and eventually banishment of the complex I inhibitor rotenone [4][5]. The question is, therefore, the risk of inhibition of human SDH following accidental or chronic (contamination of food) exposure to these SDHIs and possible consequences. Toxicological studies in rodent or other mammals were required for authorization of use of SDHIs. The blockade of cellular respiration in mammals, as with cyanide, results in death within a few minutes, if not seconds, and SDHIs would have been classified as highly toxic if that were the case. While these toxicological studies are not made public, they allowed the definition of tolerable exposure levels. The existing literature shows that other inhibitors of SDH result in the poisoning of mammalian SDH, with neurological consequences that become severe (irreversible lesions with neuronal death) when inhibition of SDH reaches 30–50% [6]. Genetic data obtained in SDHD-KO mice [7], and with alleles causing a loss of function of SDH in humans that are autosomal and recessive [8][9], indicate that SDH activity is required. Transient inhibition of cellular respiration is not inevitably deleterious in the mid-term, and transient ischemia, or inhibition of cellular respiration (preconditioning), including with an SDH inhibitor [10][11], triggers adaptive mechanisms that protect against a subsequent higher intensity event of the same nature. This opens the possibility that a given inhibitor of mitochondrial activity would show a biphasic effect with a rather positive side for low levels of exposure and deleterious consequences for the higher levels. Low and high may mean, here, dosage and/or duration. Finally, too much activity of the SDH enzyme might have deleterious consequences, and partial inhibition of SDH might be considered a therapeutic approach [12].
2. SDH and Cellular Energy Metabolism
Because SDH participates in the cellular energy metabolism, the main characteristics of the latter will be briefly summarized before more detailed examination of SDH itself. Cellular energy expenditure is mainly supported by the hydrolysis of the last phosphate bond of adenosine triphosphate (ATP > ADP + Pi). ATP is regenerated continuously by the reverse reaction (phosphorylation of ADP). In mammals, this phosphorylation is the result of two processes: the mitochondrial respiration, or the lactic fermentation. The latter is actually efficient locally/transiently, but lactate is not significantly released as waste in the environment, and eventually mitochondrial respiration is the ATP provider in mammals. The respiration of an animal uses oxygen (O2) and releases carbon dioxide (CO2). It is pertinent to examine separately these two processes. CO2 comes from substrate oxidation (metabolism), which provides electrons to the oxidative phosphorylation (Oxphos) that reduces O2 into water (Figure 1). Metabolism (as defined above) includes a large number of enzymes, and few of them release ATP during carbon oxidation. However, Oxphos is quantitatively the main contributor to ATP regeneration. Oxphos is ensured by five enzymatic complexes and two electron shuttles (quinone and cytochrome-c), all located in the inner mitochondrial membrane. Complexes I-IV constitutes the mitochondrial respiratory chain (MRC) that catalyze successive steps of electron transfer from a donor to oxygen. Complexes I, III, and IV convert the energy of oxidation into a proton electrochemical gradient used by complex V; that is the ATP producing machine. Complex II is the SDH.
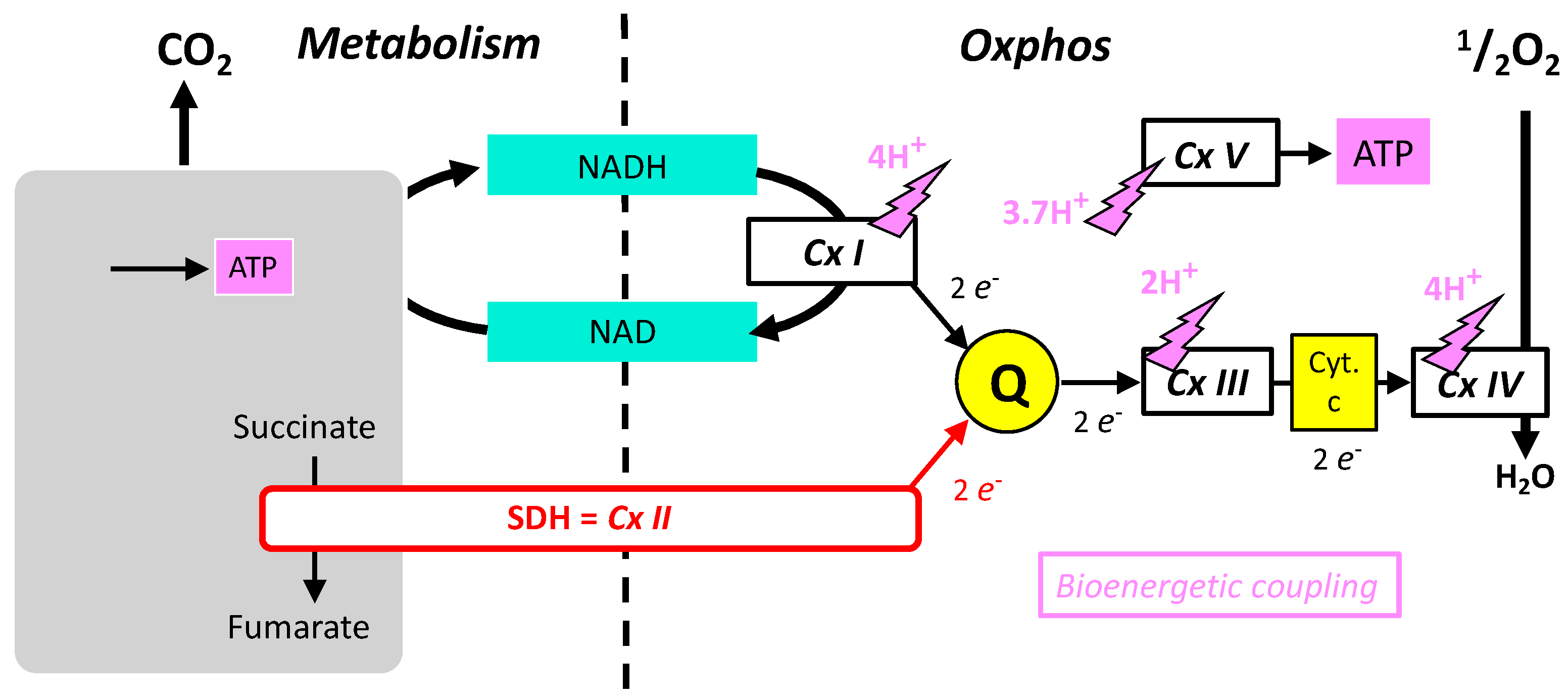
Figure 1. Schematic representation of the role of SDH and its relationships with oxidative metabolism (Metabolism), shown as a grey box releasing CO2 and producing some ATP and oxidative phosphorylation with the five complexes (CxI-V) and redox shuttles (NAD/NADH) quinone (Q) and cytochrome-c.
Comparison between SDH/Complex II and the Other Mitochondrial Respiratory Complexes
The similarities, relations, and differences between SDH/complex II and other respiratory complexes are summarized below:
-
Respiratory complexes are very large heteromeric assemblies (more than one million dalton) inserted in the mitochondrial inner membrane. This means a complex/long assembly process. A consequence is a long lifetime that allows integration and binding of inhibitors with time. The large majority of the subunits constituting these respiratory complexes are encoded by the nuclear genome, but in addition complexes I, III, IV and V contain a minority of subunits coded by the mitochondrial genome. In contrast, all subunits of complex II are coded by the nuclear genome.
-
The oxidative phosphorylation mechanism follows Mitchell’s chemiosmotic theory, according to which proton gradient and movement explain the bioenergetic coupling (Figure 1). Accordingly, complexes I, III, IV, and V associate proton movement to another reaction: redox reactions to proton pumping in complexes I, III, and IV, or phosphorylation of ADP into ATP to proton reentry in complex V. In contrast, complex II does not catalyze proton movement and is uniquely a redox enzyme.
-
Complexes I, II, III, and V could operate in the opposite direction depending on forces, which for complexes I, III, and V include the proton gradient (membrane potential). The proton gradient has no direct effect on complex II (SDH) that (in absence of inhibitors) is uniquely sensitive to the ratio of succinate/fumarate and to the reduction/oxidation state of quinone.
-
Complexes I, III, and IV of the MRC accept electrons from electron shuttles (NAD, quinone, cytochrome c) and, therefore, the link with metabolic redox reactions is indirect. In contrast, the link between carbon metabolism and electron supply to the respiratory chain is direct and relies on electron transfer within the SDH itself.
-
Oxidation of glucose and of fatty acids loads the largest part of the electrons on the NAD/NADH shuttle and complex I is, therefore, the main entry for electrons in the respiratory chain. However, complex II and the electron-transferring flavoprotein complex reoxidizing FADH2 from beta oxidation of fatty acids and a few others yield electrons directly to quinone with two consequences: (i) the yield in ATP of the electron transfer to oxygen is lower (1.6 ATP) per two electrons (oxygen atom) instead of 2.7 with complex I, and, (ii) quinone appears to be the point of convergence for the entry of electrons, with only one exit (complex III) and, consequently, competition between electron donors could take place, notably between complex I and II.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24044045
References
- Sierotzki, H.; Scalliet, G. A review of current knowledge of resistance aspects for the next-generation succinate dehydrogenase inhibitor fungicides. Phytopathology 2013, 103, 880–887.
- Coles, C.J.; Singer, T.P.; White, G.A.; Thorn, G.D. Studies on the binding of carboxin analogs to succinate dehydrogenase. J. Biol. Chem. 1978, 253, 5573–5578.
- Bénit, P.; Kahn, A.; Chretien, D.; Bortoli, S.; Huc, L.; Schiff, M.; Gimenez-Roqueplo, A.-P.; Favier, J.; Gressens, P.; Rak, M.; et al. Evolutionarily conserved susceptibility of the mitochondrial respiratory chain to SDHI pesticides and its consequence on the impact of SDHIs on human cultured cells. PLoS ONE 2019, 14, e0224132.
- Available online: http://www.epa.gov/oppsrrd1/reregistration/REDs/rotenone_red.pdf (accessed on 20 December 2022).
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306.
- Brouillet, E.; Guyot, M.-C.; Mittoux, V.; Altairac, S.; Condé, F.; Palfi, S.; Hantraye, P. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J. Neurochem. 1998, 70, 794–805.
- Piruat, J.I.; Pintado, C.O.; Ortega-Sáenz, P.; Roche, M.; López-Barneo, J. The mitochondrial SDHD gene is required for early embryogenesis, and its partial deficiency results in persistent carotid body glomus cell activation with full responsiveness to hypoxia. Mol. Cell Biol. 2004, 24, 10933–10940.
- Pagnamenta, A.T.; Hargreaves, I.P.; Duncan, A.J.; Taanman, J.-W.; Heales, S.J.; Land, J.M.; Bitner-Glindzicz, M.; Leonard, J.V.; Rahman, S. Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol. Genet. Metab. 2006, 89, 214–221.
- Van Coster, R.; Seneca, S.; Smet, J.; Van Hecke, R.; Gerlo, E.; Devreese, B.; Van Beeumen, J.; Leroy, J.; De Meirleir, L.; Lissens, W. Homozygous Gly555Glu mutation in the nuclear-encoded 70 kDa flavoprotein gene causes instability of the respiratory chain complex II. Am. J. Med. Genet. A 2003, 120A, 13–18.
- Bracko, O.; Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Tavazzi, B.; Artmann, J.; Wong, E.C.; Buxton, R.B.; Weller, M.; Luft, A.R.; et al. 3-Nitropropionic acid-induced ischemia tolerance in the rat brain is mediated by reduced metabolic activity and cerebral blood flow. J. Cereb. Blood Flow Metab. 2014, 34, 1522–1530.
- Riepe, M.W.; Niemi, W.N.; Megow, D.; Ludolph, A.C.; Carpenter, D.O. Mitochondrial oxidation in rat hippocampus can be preconditioned by selective chemical inhibition of succinic dehydrogenase. Exp. Neurol. 1996, 138, 15–21.
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515.
This entry is offline, you can click here to edit this entry!