Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Obeticholic acid (OCA) or 6-alpha-ethyl-chenodeoxycholic acid is a semisynthetic modified bile acid derivative that acts on the farnesoid X receptor (FXR) as an agonist with a higher potency than bile acid. The FXR is a nuclear receptor highly expressed in the liver and small intestine and regulates bile acid, cholesterol, glucose metabolism, inflammation, and apoptosis.
- obeticholic acid
- non-alcoholic fatty liver disease
- metabolic syndrome
1. Introduction
The farnesoid X receptor (FXR) is a nuclear receptor that can form a heterodimer with the retinoid X receptor (RXR) or bind to its gene response element in its monomeric form. The binding to the FXR element (FXRE) can result in differential activation or repression of downstream targets [1,2,3,4,5,6]. The FXR is a nuclear receptor highly expressed in the liver and small intestine, especially the distal ileum, as well as the kidneys, adrenal glands, muscles, adipose tissue, and cardiac muscle, that regulates bile acid, cholesterol, glucose metabolism, inflammation, and apoptosis [1,2,3,4,6,7]. The role of FXR in liver and small intestine has been extensively studied in regulating bile acid, cholesterol, glucose metabolism, inflammation, and apoptosis [8]. In the liver, FXR primarily acts as a bile acid sensor. FXR activation causes release of FGF19, acts as an endocrine signaling molecule from enterocytes to the liver, which causes a decrease in bile acid synthesis and release. LRH1 and SHP levels are increased due to FXR activation, which also decreases bile acid synthesis via inhibition of CYP7A1, cholesterol utilization and fatty acid metabolism. Several animal and clinical studies are underway to target therapies, such as Obeticholic acid (OCA), towards FXR for the treatment of cardiovascular disease, male infertility, kidney failure, obesity, and vascular disease [9,10,11]. Obeticholic acid (OCA) or 6-alpha-ethyl-chenodeoxycholic acid is a semisynthetic modified bile acid (BA) derived from chenodeoxycholic acid that is a potent activator of the farnesoid X receptor (FXR) [1,2,3,4,12]. Figure 1 shows the effects of OCA on physiology and pathology where it crosses the cell membrane in liver and enterocytes to activate FXR that can form a heterodimer with the RXR, homodimer with FXR, or bind to DNA as a monomer.The FXR is a nuclear receptor highly expressed in the liver and small intestine that regulates bile acid, cholesterol, glucose metabolism, inflammation, and apoptosis. Several animal and clinical studies are underway for targeted therapies towards FXR for the treatment of cardiovascular disease, male infertility, kidney failure, obesity, and vascular disease [9,10,11].
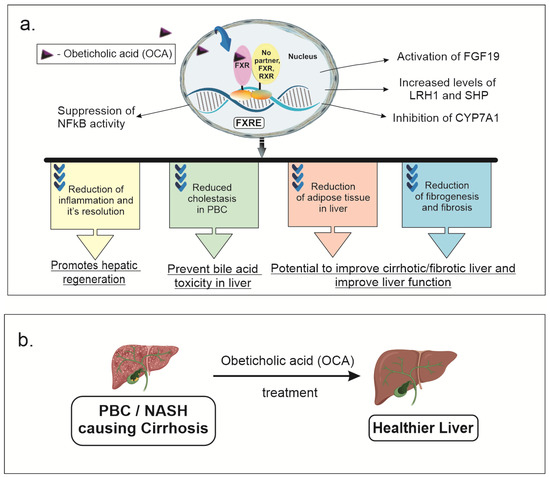
Figure 1. OCA effects on physiology and pathology: (a) OCA crosses the cell membrane in liver and enterocytes to activate FXR (FXR) that can form a heterodimer with the retinoid x receptor (RXR), a homodimer with FXR, or bind to DNA as a monomer. This can activate various signaling pathways, decreasing bile acid synthesis, fatty acid and cholesterol metabolism, glucose metabolism, inflammation and fibrosis. FXR activation causes release of fibroblast growth factor 19 (FGF19) which acts as an endocrine signaling molecule released from enterocytes to the liver causing decreased bile acid. Liver related homolog 1 (LRH1) and small heterodimer protein (SHP) levels are increased due to FXR activation, which also decreased bile acid synthesis via inhibition of CYP7A1, cholesterol utilization and fatty acid metabolism. This all improves cholestasis in patients with PBC. In mouse models, anti-inflammatory effects of OCA are mediated via suppression of nuclear factor k activator of B cells (NFkB) signaling. This mediates reduction in hepatic inflammation and fibrosis seen in mouse models of non-alcoholic steato hepatitis (NASH). (b) Improvement in liver functional status after OCA treatment in mouse models of cirrhosis due to NASH or primary biliary cholangitis (PBC).
2. The Effects of OCA on Different Physiological Processes through FXR Activation
2.1. OCA Effect on Bile Acid Synthesis
One of FXR’s main roles is to inhibit bile acid synthesis through regulating the expression of small heterodimer partner (SHP). SHP inactivates liver-related homolog-1 protein (LRH-1), which in turn represses the expression of the cholesterol 7α-hydroxylase (CYP7A1) enzyme [13]. CYP7A1 is the rate-limiting step in BA synthesis within the liver. Another mechanism by which FXR can downregulate CYP7A1 is via the induction of fibroblast growth factor 19 (FGF19) within enterocytes located in the ileal region. This molecule can act as an endocrine signaling hormone, activating the JNK signaling cascade, and causing a downregulation of CYP7A1 expression after reaching the liver via enterohepatic circulation [14,15]. Through inhibiting and downregulating CYP7A1, FXR is believed to exert hepatoprotective properties by preventing the accumulation of toxic BA buildup in the liver and promoting hepatic regeneration after liver damage in mouse models [16,17,18].
The primary method by which OCA reduces circulating bile salts is through upregulation of the bile salt exporter pump (BSEP) to increase bile salt excretion into the biliary tree [19,20,21]. In addition, OCA decreases intestinal cholesterol absorption rather than increasing biliary cholesterol production [22]. OCA also inhibits hepatic sterol 12 hydroxylase (CYP8B1) and cholesterol 7 hydroxylase (CYP7A1), in part by inducing a small heterodimer partner [22]. This results in a smaller bile acid pool and a change in the composition of the bile, with an increase in the proportion of α/β-muricholic acid and a decrease in taurocholic acid [22]. Additional studies using primary hepatocytes showed that OCA treatment suppressed bile acid synthesis genes (CYP7A1, CYP27A1) and increased bile efflux genes (ABCB4, ABCB11, OSTA, OSTB) [23,24].
2.2. OCA Effect on Fatty Acid Metabolism
In addition, the FXR plays an important role in fat metabolism, which includes regulating the expression of several lipoproteins, including apolipoprotein E and very low-density lipoprotein receptor (VLDLR) [25,26,27,28,29,30]. By phosphorylating insulin responsive substrate-1 on serine 312 in the liver and muscles, FXR activation was found to reduce body weight gain, prevent fat from accumulating in the liver and muscles, and reverse insulin resistance [26]. Genes involved in fatty acid production, lipogenesis, and gluconeogenesis had their liver expression decreased by FXR activation. Free fatty acid production in the muscles was also decreased by FXR therapy [26]. A subsequent study using OCA reduced the amount of visceral adipose tissue, steatosis, and other inflammatory markers in rabbits fed a high fat diet [31]. Similar results were also observed in mice, hamsters, and human [32,33,34,35].
2.3. OCA’s Effect on Vascular and Inflammatory Processes in the Liver
Within vascular smooth muscle cells, OCA can induce smooth muscle cell death and downregulate interleukin (IL)-1β-induced inducible nitric oxide synthase and cyclooxygenase-2 expression [36]. In addition, OCA suppressed smooth muscle cell migration stimulated by platelet-derived growth factor-BB. However, a recent mouse model study suggested that OCA may not help with hepatic regeneration resulting from liver injury [37]. Further studies will need to be done to assess whether OCA may revert liver damage in human patients. The binding of OCA to FXR has also been shown to decrease the expression of nuclear factor κ light-chain enhancer of activated B cells (NF-κB) by inducing expression of cytochrome P450. This process can reduce inflammasome formation and prevent the development of various liver and vascular pathologies [38].
Along with its effects on metabolism, OCA also acts as an anti-inflammatory agent via activation of FXR in liver [36,39,40,41,42,43,44]. Several animal studies have shown that loss of FXR or FXR deficiency, results in increased hepatic inflammation and fibrosis [39,40,41,42,45,46]. Specifically, it was found that activation of FXR reduced the production of fibrosis from hepatic stellate cells through altering the activities of peroxisome proliferator-activated receptor gamma (PPARγ) and SHP. In response, there is a corresponding reduction in the pro-fibrotic activities of α1 collagen, TGF-β1, and NLR family pyrin domain containing 3 (NLRP3) inflammasome activation [43,46]. Further improvements in hepatic inflammation and fibrosis were also observed when OCA was combined with glucagon-like peptide-1 receptor (GLP-1R) agonists [47]. A similar result was also observed when OCA was combined with an anti-apoptosis inhibitor (IDN-6556), PPAR-α/δ agonists, and statins [48,49,50]. Further studies using primary hepatocytes treated with OCA reduced TGFβ and IL-6 signaling pathways as well as reduced HDL genes (SCARB1, ApoAI, LCAT) while LDL genes (ApoB, CYP7A1) increased [23].
In recent years, the FXR group of bile acid receptors is currently under investigation for its potential role in the treatment of primary biliary colangitis (PBC), non-alcoholic steatohepatitis (NASH), non-alcoholic fatty liver disease (NAFLD), and primary sclerosing cholangitis (PSC) [51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71]. In 2016, the Food and Drug Administration approved OCA for treating PBC refractory to ursodeoxycholic acid (UDCA) [45]. Besides OCA, several other FXR agonists are under investigation for the treatment of PBC and NASH [72,73,74,75]. Recent clinical studies suggest OCA may work synergistically with lipid modifying medications to further improve long-term outcomes with primary sclerosing cholangitis. Specifically, OCA improves morbidity and mortality in NASH patients with their different histological, metabolic, and biochemical issues in PBC, PSC, or liver patients.
This entry is adapted from the peer-reviewed paper 10.3390/futurepharmacol3010017
This entry is offline, you can click here to edit this entry!