Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemical Research Methods
Sulfane sulfurs, which include hydropersulfides (RSSH), hydrogen polysulfides (H2Sn, n > 1), and polysulfides (RSnR, n > 2), play important roles in cellular redox biology and are closely linked to hydrogen sulfide (H2S) signaling. While most studies on sulfane sulfur detection have focused on sulfane sulfurs in the whole cell, increasing the recognition of the effects of reactive sulfur species on the functions of various subcellular organelles has emerged, such as mitochondria. This has driven a need for organelle-targeted detection methods.
- sulfane sulfur
- fluorescent probe
- organelle
1. Introduction
Biological sulfane sulfurs (S0), including hydropersulfides (RSSH), polysulfides (RSSnSR), hydrogen polysulfides (H2Sn, n ≥ 2), and protein-bound elemental sulfurs (S8), have become increasingly recognized as important reactive sulfur species (RSS) with distinct functions in redox biology that are closely linked to hydrogen sulfide (H2S) signaling [1]. Sulfane sulfurs are sulfur atoms with six valence electrons and no charge that are covalently bonded to other sulfur atoms. Significantly, sulfane sulfurs have been discovered to influence various physiological and pathological processes, including activating the transient receptor potential ankyrin 1 (TRPA1) channel, relaxing vascular smooth muscles, mediating neurotransmission, and regulating inflammation [2,3,4,5]. Yet, due to their instabilities, sulfane sulfurs such as RSSH and H2Sn are understudied despite their active involvement in redox signaling. Considering the importance of sulfane sulfurs in biological systems, the development of detection methods for these species is important to better understand their biological mechanisms of action and potential therapeutic applications.
Some of the most popular detection methods for sulfane sulfurs or other biologically important analytes are fluorescence spectroscopy and fluorescence microscopy [6,7,8,9,10,11,12,13,14,15]. These methods involve the usage of fluorescent probes, which are important tools in the study of biological systems because they allow researchers to visualize and track specific molecules or processes within cells and tissues. By emitting light when excited by a specific wavelength of light, fluorescent probes allow scientists to detect and even quantify the presence of specific molecules in real time [13,14,15]. Thus, fluorescent probes can answer fundamental questions regarding the production and mechanisms of action for sulfane sulfurs in biological samples, making these probes essential for medical diagnosis, treatment, and basic biomedical research.
Most reported fluorescent probes and studies for sulfane sulfur detection examine cellular sulfane sulfur levels rather than those in subcellular microenvironments. Yet, organelles are specialized subunits within cells that perform specific functions that are essential for the overall health and survival of the cell. In events of stress or malfunction, disease can result. For example, the mitochondria are involved in many critical processes, including the regulation of cell signaling and differentiation, cell death pathways, and the cell cycle [16]. Mitochondrial oxidative damage has been found to contribute to a wide range of human disorders, including ischemia-reperfusion injury and aging-associated dysfunction [17]. While studies have found that H2S offers cardioprotective effects by preserving mitochondrial function, sulfane sulfurs are less well-studied. It has been reported, however, that the majority of bound sulfane sulfurs in cells are in the mitochondria, suggesting the importance of this organelle in maintaining cardiovascular homeostasis [18]. It is also known that the mitochondrial enzyme sulfide quinone oxidoreductase (SQOR) rapidly converts H2S into sulfane sulfurs (persulfides and polysulfides) which are then stored in the mitochondria until they are released in response to physiological signals [19]. Considering that this organelle has been found to play key roles in diseases and sulfane sulfurs have been found as actual signaling species in a range of biological activities previously attributed to H2S, an accurate, sensitive, and real-time method for detecting sulfane sulfurs in the mitochondria is essential to understand their mechanisms.
Other subcellular organelles also have specific functions that contribute to the operation of a cell and can result in disease in the event of dysfunction. For example, lysosomes are single-layered membrane organelles that are responsible for cellular waste digestion and contain acidic environments and hydrolases. RSS plays a role in the regulation of lysosomal activity and membrane permeability, thus affecting many biological processes [20]. The rough endoplasmic reticulum (ER) is responsible for protein synthesis, while the smooth ER is primarily involved in calcium signaling, lipid synthesis, and carbohydrate metabolism. ER stress and protein misfolding have been associated with diseases, including myocardial ischemia-reperfusion (MI/R) injury, cardiomyopathy, heart failure, hypertension, and diabetes [21,22,23,24]. The Golgi apparatus processes, packages, and transports proteins and lipids. The lysosomes, ER, and Golgi apparatus have connected functions as part of a secretory pathway with the cell membrane; therefore, the subcellular targeting of sulfane sulfurs in these organelles using fluorescent probes is critical for better understanding the physiological and pathological impacts of sulfane sulfurs on various diseases. This greater knowledge may even have potential implications for clinical diagnosis and improved therapeutics.
Due to the rising interest in organelles, sulfane sulfurs, and the role of sulfane sulfurs in maintaining intracellular redox homeostasis, developments in organelle-targeted fluorescent probes for sulfane sulfurs have been made in recent years.
2. Mitochondria-Targeting Probes
Mitochondria are the major source of reactive oxygen species (ROS). During mitochondrial respiration, nearly 0.1–4% of oxygen is reduced to the superoxide ion (O2•−) due to electron leakage from the respiratory chain. This species is then transformed into other ROSs via enzymatic or non-enzymatic pathways [25]. Meanwhile, endogenously produced H2S is oxidized in the presence of mitochondrial ROS to form sulfane sulfurs, which can also be formed directly via enzymes such as 3MST. Thus, to better understand redox homeostasis, monitoring sulfane sulfurs via fluorescence imaging is useful. Mitochondria possess a unique double-layered membrane structure with a negative membrane potential (as high as −180 mV) [26]. Hence, in most cases, mitochondria-targeted probes possess at least one lipophilic cation [27]. Non-cationic probes can be functionalized by attaching triphenyl phosphonium [20] or pyridinium [28,29,30] as the anchor. However, functionalized cationic dyes are also known to target other organelles [31,32,33,34,35]. Based on colocalization experiments with commercially available mitochondria-targeting dyes, some non-cationic dyes have been reported to selectively target the mitochondria due to their unique structures [36].
In 2015, Chen and coworkers developed a reaction-based near-infrared (NIR) fluorescent probe (Mito-ss) for the detection of mitochondrial hydrogen polysulfides (H2Sn, n > 1) [37]. Mito-ss consists of (i) a NIR dye based on the azo-BODIPY chromophore, (ii) a lipophilic triphenyl phosphonium group, and (iii) an H2Sn-reactive nitrofluorobenzoate moiety (Scheme 1a). They chose a NIR fluorophore because NIR lights possess certain advantages, including deep tissue penetration, low cytotoxicity, and minimum background noise. Nitrofluorobenzoate is a commonly used functional group for the design of H2Sn sensors [38]. Nitrofluorobenzoate bears two electrophilic sites. H2Sn first reacts with it via nucleophilic aromatic substitution (SNAr) to replace the F atom and form a persulfide (-SSH) intermediate, which then undergoes a spontaneous intramolecular cyclization with the ester group to uncage the fluorophore. Due to its electron with-drawing nature, nitrofluorobenzoate quenches the fluorescence of the azo-BODIPY chromophore via a donor-excited photoinduced electron transfer (d-PET) process. As such, Mito-ss is a reaction-based ‘turn-on’ sensor for H2Sn. Mito-ss reacts rapidly (~30 s) with H2Sn and exhibits a 24-fold fluorescence increase at an emission of 730 nm. The probe was examined with various ROS, reactive nitrogen species (RNS), and other RSS and demonstrated no fluorescence turn-on. Biothiols such as glutathione (GSH), cysteine (Cys), N-acetyl-L-cysteine, etc., could react with Mito-ss. However, as the reaction stopped at the SNAr step, no fluorescence was observed. The limit of detection (LOD) for Mito-ss was calculated to be 25 nM. The probe was used for the real-time detection of exogenous and endogenous H2Sn using six different cell lines. Mito-ss was also found to be suitable for the in vivo detection of exogenously injected H2Sn in BALB/c mice.
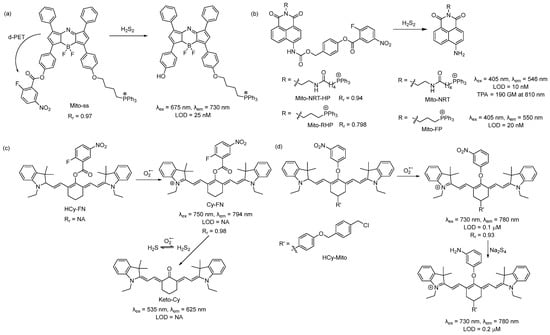
Scheme 1. Structures and reactions of probes (a) Mito-ss, (b) Mito-NRT-HP, (c) HCy-FN, and (d) HCy-Mito.
Using the same nitrofluorobenzoate reaction site, Han et al. developed a ratiometric fluorescent probe, Mito-NRT-HP, for the detection of mitochondrial H2Sn in 2019 [39]. The structure of Mito-NRT-HP is similar to Mito-ss, though with a two-photon responsive naphthalimide fluorophore instead of a single-photon responsive fluorophore. The naphthalimide fluorophore has the advantage of easily tunable photophysical properties by blocking and/or unblocking the internal charge transfer (ICT) process. It is highly photostable, resistant to pH interference, and possesses a large two-photon absorption cross-section. Importantly, 1,8-naphthalimide can be easily functionalized by simple synthetic tailoring [40]. The main advantages of two-photon excitation over single-photon excitation include deep tissue penetration, lesser damage, poor scattering, etc. In the case of two-photon excitation, a femto second pulsed laser is used, and the molecule can be excited only at the focal point of the laser. Three-dimensional imaging can be obtained [41]. Upon titrating with different concentrations of Na2S2, it was found that Mito-NRT-HP gave ratiometric responses with a changing fluorescence color from blue to green. When the solution of Mito-NRT-HP was treated with H2Sn, the initial emission maximum at 478 nm decreased gradually, with a concomitant peak increase at 546 nm. The detection limit was 10 nM which suggests that Mito-NRT-HP could have the relevant sensitivity needed for the quantitative detection of H2Sn under physiological conditions. The two-photon absorption cross-section values (δ) of Mito-NRT-HP and its fluorophore Mito-NRT (Scheme 1b) were recorded in a buffer using a pulsed laser, and fluorescein was used as the reference molecule. Their δ values were measured over a range of wavelengths starting from 750 nm to 825 nm. The highest δ was 290 GM [1 GM (Goeppert-Mayer) = 10−50 cm4 s photon−1] for Mito-NRT-HP and ~190 GM for Mito-NRT at 810 nm. Mito-NRT-HP was found to exhibit good cell permeability and weak cytotoxicity, which was suitable for the ratiometric imaging of endogenous H2S2 in cells. Mito-NRT-HP was colocalized with MitoTracker Red (MTR) and LysoTracker Red (LTR), and the colocalization coefficients were found to be 0.94 and 0.42, respectively, indicating that Mito-NRT-HP was specifically localized in the mitochondria. Using two-photon microscopy, images of the tissue slices from mice with lipopolysaccharide (LPS)-induced acute organ injury were taken and compared with the control tissues. The enhanced fluorescence in the former case was observed. In 2021, Han et al. reported a similar probe for the detection of mitochondrial H2Sn during H2O2-induced redox imbalance [42]. The structure of this probe (Mito-RHP) only differed from Mito-NRT-HP in the linker between naphthalimide and the triphenylphosphonium unit. Upon the addition of Na2S2 to the solution of Mito-RHP, the initial emission spectra of Mito-RHP at 485 nm gradually decreased, and a continuous increase in the new peak to 550 nm was observed, along with a change in fluorescence color from blue to yellowish green. In this case, the Stokes shift was 109 nm, which was higher than that of Mito-NRT-HP. The detection limit was calculated to be 20 nM. Other properties, such as photostability, solubility, permeability, and cytotoxicity, were similar. However, the mitochondria-targeting ability of the new probe (overlap coefficient = 0.836) was not as good as that of Mito-NRT-HP (overlap coefficient = 0.94). The in vivo imaging of exogenous H2Sn (using Na2S2) was performed in zebrafish using Mito-RHP.
An interesting single-component multi analyte responsive NIR fluorescent probe was reported by Chen and coworkers in 2015 for the detection of the superoxide ion (O2•−) and H2Sn to understand redox homeostasis in the mitochondria [43]. Both O2•− and H2Sn are short-lived reactive species, and their concentrations change quickly. To solve this problem, they developed a cyanine-based NIR probe, HCy-FN. This probe consists of two different reaction sites: one for the abstraction of hydrogen to detect O2•− and the other for the detection of H2Sn using nitrofluorobenzoate (Scheme 1c). Both sensing steps were monitored by two different channels. Upon reacting with O2•−, HCy-FN was oxidized to Cy-FN, and this transformation was monitored by an increase in the emission intensity from channel 1 at 794 nm (λex = 750 nm). Next, the nitrofluorobenzoate part of Cy-FN reacted with H2Sn to result in a decrease in the emission intensity of channel 1 followed by an increase in the emission intensity in channel 2 at 625 nm (λex = 535 nm) due to the formation of Keto-Cy. They examined different ROS with HCy-FN and found that only O2•− was able to oxidize the probe. Similarly, the reactivity of other RSS towards Cy-FN was also evaluated, and no changes in emission spectra were noted. HCy-FN was used for the detection of exogenous and endogenous H2Sn with the macrophage cell line RAW264.7 to monitor both sensing steps by dual channel emission. It was found that the Pearson correlation coefficient (Rr) of Cy-FN and mitochondria-localizing Rhodamine 123 was 0.98, confirming that Cy-FN was localized in the mitochondria. Moreover, HCy-FN could detect endogenously produced O2•−/H2Sn in BALB/c mice. This work represents an interesting way to detect O2•−/H2Sn in the biological system. However, the claim that the probe is capable of monitoring mitochondrial O2•−/H2Sn may not be accurate. The authors only provided the Rr value for the intermediate compound Cy-FN and not for the actual probe. The structure of HCy-FN suggests that it may not be a suitable candidate to target the mitochondria because of the lack of a lipophilic cationic moiety.
In 2016, Chen and coworkers developed a probe (HCy-Mito) for the selective detection of superoxide anion (O2•−) and H2Sn in the mitochondria [44]. The reaction sites for O2•− and H2Sn were the reduced cyanine dye (similar to HCy-FN) and m-nitrophenyl ether (Scheme 1d). In the presence of O2•−, Hcy-Mito was oxidized to form a cyanine derivative, and the reduced nature of H2Sn converted the nitro group to -NH2, which terminated the d-PET process and resulted in an increase in emission intensity to 780 nm. The detection limits for O2•− and H2Sn by HCy-Mito were found to be 0.1 μM and 0.2 μM, respectively. In vitro experiments with RAW264.7 cells by HCy-Mito suggest that it could image exogenous and endogenous O2•−/H2Sn and localize specifically in the mitochondria (Rr = 0.93). This probe was further utilized for the in vivo detection of O2•− (generated from phorbol myristate acetate (PMA) and H2Sn (via injected Na2S4) in BALB/c mice.
In 2019, Meng et al., used a different reaction site based on the 2-(acylthio)benzoate for the design of a mitochondria-targeted probe for H2Sn [45]. This template utilized both the nucleophilic and electrophilic nature of H2Sn for its recognition (Scheme 2a) [46]. Briefly, the thioester exchange between the H2Sn and 2-(acylthio)benzoate produced a thiophenol derivative, which, in turn, reacted with H2Sn to form an -SSH intermediate. This intermediate underwent an intramolecular cyclization to release the fluorophore. This template was attached to a red-emitting fluorophore to develop the probe, HQO-PSP. HQO-PSP itself was non-fluorescent but, upon sensing H2Sn, exhibited a fluorescence turn-on (86-fold) at an emission of 633 nm due to the formation of the keto derivative HQO. The probe was found to be relatively fast (7 min) and highly selective to H2Sn, with a detection limit of 95.2 nM. In vitro studies with A549 cells revealed that HQO-PSP could specifically localize within the mitochondria (Rr = 0.98) and selectively image exogenously added H2Sn in the live cells.
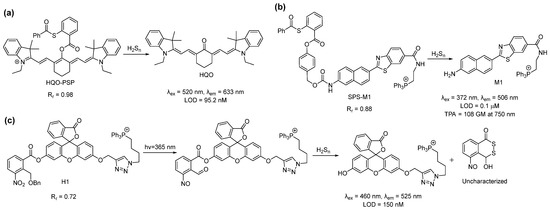
Scheme 2. Structures and reactions of probes (a) HQO-PSP, (b) SPS-M1, and (c) H1.
In the same year, Choi et al. reported that the ratiometric probe SPS-M1 for mitochondrial H2Sn detection was based on a two-photon excitable naphthalene fluorophore [47]. The reaction site was the same as that of HQO-PSP except for an additional self-immolating carbamate linker (Scheme 2b). The probe exhibited a blue fluorescence (λem = 429 nm) but produced the deprotected yellow fluorescent dye M1 (λem = 506 nm) upon sensing H2Sn. Interestingly, the two-photon absorption (TPA) cross-section (δ) of SPS-M1 and M1 was found to be 11 and 108 GM, respectively, at 750 nm. The large TPA cross-section resulted from the strong ICT process in M1. SPS-M1 was found to be suitable for the quantification of H2Sn in live cells, and the in vitro detection limit was 1 μM. The two-photon microscopic imaging with SPS-M1 for endogenous H2Sn using the wild-type and Parkinson’s disease (PD) model neurons and brain tissues of mice revealed that H2Sn concentrations were higher in the PD model.
Another interesting approach for the spatiotemporal detection of mitochondrial H2Sn was reported by Han et al. in 2018 [48]. Probe H1 consisted of a fluorescein dye attached to a triphenylphosphonium group and a nitrobenzyl photoactivable protecting group (Scheme 2c). Upon irradiation with UV light (365 nm), the nitrobenzyl part produced an aldehyde derivative, which served as the H2Sn recognition site. H2Sn attacks the aldehyde group to form a persulfide intermediate, which then should undergo cyclization to liberate the fluorophore and generate a side product (4-hydroxybenzo[d][1,2]dithiin-1(4H)-one). H1 showed a turn-on of fluorescence at 525 nm only when it was irradiated with UV light along with H2S2 in the solutions. The detection limit was calculated to be 150 nM. The targeting ability of H1 was confirmed by counterstaining with MitoTracker Green (MTG) (Rr = 0.72). Although this photo-triggered probe was interesting, the authors did not provide experimental support for the proposed detection mechanism. This aldehyde-based intermediate may also possess some problems as 2-formyl carboxylate is a well-known H2S recognition site [49,50], and the aldehyde group has a high reactivity towards free cysteine [51,52,53].
This entry is adapted from the peer-reviewed paper 10.3390/antiox12030590
This entry is offline, you can click here to edit this entry!