Glucose uptake in the brain decreases because of normal aging but this decline is accelerated in Alzheimer’s disease (AD) patients. In fact, positron emission tomography (PET) studies have shown that metabolic reductions in AD patients occur decades before the onset of symptoms, suggesting that metabolic deficits may be an upstream event in at least some late-onset cases. A decrease in the availability of glucose content induces a considerable impairment/downregulation of glycosylation, which is an important post-translational modification. Glycosylation is an important and highly regulated mechanism of secondary protein processing within cells and it plays a crucial role in modulating the stability of proteins, as carbohydrates are important in achieving the proper three-dimensional conformation of glycoproteins. Moreover, glycosylation acts as a metabolic sensor that links glucose metabolism to normal neuronal functioning. All the proteins involved in β-amyloid (Aβ) precursor protein metabolism have been identified as candidates of glycosylation highlighting the possibility that Aβ metabolism could be regulated by their glycosylation. Within this framework, the present review aims to summarize the current understanding on the role of glycosylation in the etiopathology of AD, emphasizing the idea that the glucose metabolic pathway may represent an alternative therapeutic option for targeting AD. From this perspective, the pharmacological modulation of glycosylation levels may represent a ‘sweet approach’ to treat AD targeting new mechanisms independent of the amyloid cascade and with comparable impacts in familial and sporadic AD.
- Alzheimer’s disease
- neurodegeneration
- post-translational modifications
- glycans
- glycosylation
- phosphorylation
- β-amyloid
- hexosamine biosynthetic pathway
- α-secretase
- β-secretase
- γ–secretase
- Neprilysin
1. Definition
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by a progressive decline in memory, attention and language, and histopathologically the deposition of extracellular Aβ plaques and intracellular neurofibrillary tangles (NFTs) are considered the two iconic neuropathological hallmarks of AD at post-mortem.
2. Biosynthesis and Modulation of O-GlcNAcylation
Contrary to the complex N- and O-linked glycosylation, which requires several enzymes, O-GlcNAcylation is regulated by the concerted action of only two enzymes: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). For a detailed description of OGT and OGA substrate recognition see Joiner et al., 2019 [1]. OGT catalyzes the transfer of GlcNAc onto Ser and Thr residues employing Uridine 5′-diphospho-N-acetylglucosamine (UDP-GlcNAc) as a substrate, whereas OGA is the enzyme that removes O-GlcNAc from proteins [2][3]. The complete schematic representation of O-GlcNAcylation and phosphorylation is shown in Figure 1. OGT is mainly expressed in three predominant isoforms namely short OGT (sOGT), mitochondrial OGT (mOGT), and nucleocytoplasmic OGT (ncOGT). The most studied isoform is ncOGT, the primary product of ogt gene, which catalyzes the transfer of O-GlcNAc by an ordered and sequential bi-bi mechanism [4]. OGT mRNA levels are expressed in all tissues, but the highest expression has been reported in the pancreas, brain with reduced levels in other organs. Brain OGT cytosolic activity is ten times higher when compared to other tissues including muscle, heart, liver and adipose tissue [5]. OGA is expressed in two isoforms namely OGA large (OGA-L) and OGA short (OGA-S). OGA-L is localized in both the cytosol and nuclei whilst OGA-S is limited to the nucleus [6]; the latter is relatively six times less active than OGA-L in O-GlcNAc hydrolysis [7][8]. It has been demonstrated that significant OGA expression is mostly restricted to the pancreas, brain and thymus with other organs demonstrating low levels of expression [9].
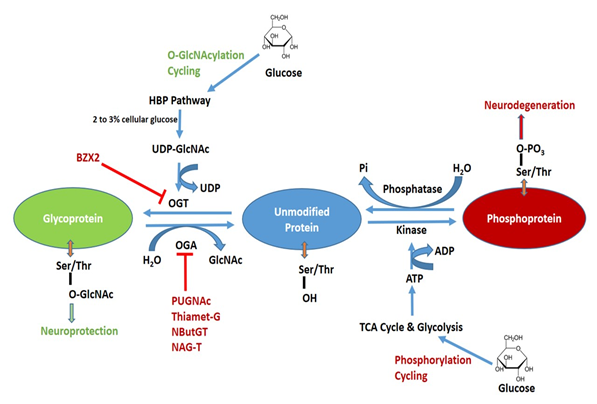
Figure 1. Schematic diagram of O-GlcNAcylation and phosphorylation processes of protein and their possible pharmacological interventions. Hexosamine biosynthetic pathway (HBP) leads to UDP-GlcNAc formation and regulates O-GlcNAcylation. O-GlcNAc transferase (OGT) is inhibited by BZX2 whilst O-GlcNAcase (OGA) is inhibited by PUGNAc, Thiamet-G, NButGT and NAG-T. These OGT and OGA inhibitors are targeted to modulated O-GlcNAc levels in neurodegenerative diseases. Tricarboxylic acid (TCA) and glycolysis cycle regulate protein phosphorylation. Kinase and phosphatase regulate phosphorylation and dephosphorylation, respectively.
The hexosamine biosynthetic pathway (HBP) is a branch of the glycolysis that is responsible for the production of UDP-GlcNAc, a key substrate for protein glycosylation. O-GlcNAc is closely regulated by cellular glucose concentrations and 2 to 3% of total glucose is transformed into UDP-GlcNAc [10]. Therefore, O-GlcNAcylation is considered a nutrient sensor and its dependence is not only confined to glucose metabolism but also influenced by amino acid (glutamine), fatty acids (acetyl-CoA) and nucleotide (uridine triphosphate) [11][12]. Thus, fluctuations in nutrition balance could alter its signaling pathway. In fact, it has been demonstrated that hyperglycemic conditions lead to an increase in O-GlcNAc [13], whereas a decreased availability of glucose content induces a considerable reduction in the O-GlcNAc levels [14].
In humans, dysregulation of O-GlcNAcylation occurs in a wide range of diseases, including cancer, diabetes, and neurodegeneration. In the latter context, the pharmacological modulation of the activities of OGT and OGA may be helpful in order to understand their role in neurodegenerative diseases. OGT and OGA inhibitors have been identified, which are able to modulate the glycosylation are listed in Table 1.
Table 1. Effects of OGT and OGA Inhibitors.
|
OGT Inhibitor: |
|||
|
Name |
Subjects |
Effects |
References |
|
BZX2 |
Tau-BiFC cells |
↑ tau phosphorylation at Ser199 and Ser396 |
[15] |
|
↑ tau aggregation |
|||
|
OGA Inhibitors: |
|||
|
O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate |
SH-SY5Y cells |
↑ O-GlcNAcylation, ↑sAPPα, |
[16] |
|
(PUGNAc) |
PC12 cells |
↓ Aβ levels |
[17] |
|
↓ Phosphorylation level of tau at Ser-199, Ser-202, Thr-205, Thr-212, Ser-214, Ser-262, and Ser396 |
|||
|
1,2-dideoxy-2'-methyl-alpha-d-glucopyranoso[2,1-d]-Delta2'-thiazoline |
Human O-GlcNAcase |
Highly selective competitive OGA inhibitor |
[18] |
|
(NAG-thiazoline) |
NIH 3T3 cells |
↑ Global GlcNAcylation levels |
[19] |
|
1,2-dideoxy-2'-propyl-alpha-D-glucopyranoso-[2,1-D]-delta2'-thiazoline |
Sprague-Dawley rats |
↑ O-GlcNAc levels |
[20] |
|
(NButGT) |
C57BL/6J mice |
No alteration in glucose tolerance and insulin signaling pathways |
[21] |
|
No insulin resistance |
|||
|
3ar,5r,6s,7r,7ar)-2-(Ethylamino)-5-(Hydroxymethyl)-5,6,7,7a-Tetrahydro-3ah-Pyrano[3,2-D][1,3]thiazole-6,7-Diol |
TAPP mice |
↑ Global GlcNAcylation levels |
[22] |
|
(Thiamet-G) |
Improves performance in the Morris water maze (MWM) test |
||
|
↓ Aβ levels |
|||
|
N-(5-(((2S,4S)-2-methyl-4-(6-fluoropyridin-2-yloxy)piperidin-1-yl)methyl)thiazol-2-yl)acetamide (LSN3316612) |
Rhesus monkey |
↑ O-GlcNAc levels |
[23] |
|
Oga knock-out mice |
↓ Tau phosphorylation |
||
|
Note: ↑: Increase; ↓: Decrease. |
|||
3. Glycosylation in Alzheimer’s Disease
Besides Aβ plaques and NFTs, additional molecular events are also related with the progression of the disease and include the increase in the reactive oxygen species (ROS) production, mitochondrial dysfunction, inflammation, and a decrease in cerebral glucose uptake/utilization [24][25][26]. To the latter regard, it is widely accepted that alterations in brain structure and function precede the signs and symptoms of the disease by many years. During normal aging, glucose uptake in the brain is decreased and this decline is strongly accelerated in Alzheimer’s patients. In this context, several 2-[18F]fluoro-2-deoxy-D-glucose (FDG) PET studies have shown that metabolic reductions in patients occur decades before the onset of AD symptoms, suggesting that metabolic deficits may be a defining upstream event in at least some late-onset AD cases [27]. These results were also confirmed in other mouse models of AD (e.g., APP/PS1 and 3 × Tg-AD mice), where reduced peripheral glucose tolerance occurs prior to AD-related neuropathology or cognitive decline [28][29][30][31]. The neurons have energy requirements and high metabolic rates and as such, their functionality is directly dependent on glucose supply and utilization, and, therefore, interruptions of the glucose supply make neurons vulnerable to damage. However, the molecular mechanisms and crosstalk between the different signaling pathways involved in the regulation of glucose metabolism and cognitive improvements, or the mechanisms by which these pathways are deregulated in AD, have not been completely identified. To this regard, more recently it has been demonstrated that Wnt signaling plays a crucial role in regulating glucose metabolism in the brain, and this pathway is downregulated in AD [61,62]. In particular, Wnt signaling promotes glucose uptake and utilization in cultured hippocampal neurons and, more interestingly, ameliorates cognitive decline in APPswe/PS1dE9 transgenic mice model AD by enhancing glucose metabolism [32][33]. Therefore, it might be promising to target Wnt pathways for neuroprotection in AD. Wnt pathway is sensitive to alterations in the glycosylation state of a cell and acts as a nutritional sensor in order to couple growth/proliferation with its metabolic status[34]. In particular, UDP-GlcNAc pathway specifically regulates Wnt signaling possibly through the global changes on the glycosylation state of Wnt receptor proteins [35].
Alteration in glucose metabolism seems to lower the UDP-GlcNAc through HBP, which alleviates, in turn, the O-GlcNAc formation [36]. The involvement of glucose metabolism in AD has also been supported by the observation that type II diabetes mellitus (T2DM) is a risk factor for, and impaired glucose metabolism correlates with dementia severity in AD [[37][38][39][40]. Moreover, it has been reported that the expression levels of glucose transporter 1 (GLUT1) and GLUT3 acting as glucose transporters were significantly decreased in the brain of AD patients and streptozotocin-treated rats together with O-GlcNAcylation, whereas the phosphorylation of tau and neurofilaments were increased [41]. Among others, β-amyloid precursor protein (APP), secretases (α-, β- and γ-secretases), neprilysin, tau, and other components of the neuron cytoskeleton are candidates for O-GlcNAcylation.
APP is an integral membrane glycoprotein and exists in both mannose-capped immature oligomannose N-glycans and mature complex N-glycans [42][43]. The APP intracellular maturation process, the translocation from the Golgi to the cell membrane and metabolism are driven by glycosylation, sulfation, and phosphorylation [43]. In this regard, the inhibition of N-glycosylation with the mannosidase inhibitors swainsonine or deoxymannojrimycin altered the subcellular translocation of APP with its retention in the Golgi, indicating that changes in APP N-glycosylation state could affect the processing pathway of this protein by determining its transport from the Golgi to the cell membrane [44]. N- glycosylation of APP is required for extracellular secretion in vitro[45]. Sialylation of APP N-glycans could drive the proteolytic cleavage of APP towards the amyloidogenic-processing pathway operated by two proteases, β-site APP cleaving enzyme-1 (BACE1) and γ-secretase complex, thereby playing a crucial role in the development of AD [72,73,76–78]. O-GlcNAcylation enhances non-amyloidogenic processing resulting in neuroprotective effects [9]. O-GlcNAcylation of APP at Thr 576 is the main regulator of Aβ trafficking and processing, making this residue a possible drug target in AD [46].
α-secretase cleaves APP within the Aβ sequence and, together with γ-secretase, generates a soluble APP intracellular domain (AICD) and a secreted sAPPα fragment, which has been reported to have neuroprotective properties [47]. In this regard, increased levels of O-GlcNAcylated APP using PUGNAc augmented α-secretase activity together with an increased level of sAPPα and a reduction of Aβ40 in SH-SY5Y neuroblastoma cells. α-secretase is a member of ‘a disintegrin and metalloprotease’ (ADAM) family and, in particular, ADAM10 is the constitutive α-secretase in primary neurons[48]. Interestingly, inactivation of ADAM10 in mice (Adam10−/−) resulted in complete suppression of α-cleavage in primary neurons enhancing Aβ42 levels [49][50]. ADAM10 is a type I transmembrane glycoprotein that cleaves several plasma membrane proteins and like other ADAMs is N-glycosylated [51]. ADAM10 has four potential N-glycosylation sites (N267, N278, N439 and N551), which contain high-mannose or complex-type glycans [51]. Individual N-glycosylation site mutants were constructed, and results demonstrated that each of the N-glycosylation sites from ADAM10 is required for full-enzyme activity [51]. Further investigation will be required elucidating the precise functional role played by ADAM10 N-glycosylation in AD.
BACE1, (also known as β-secretase) cleaves APP and is a crucial protease for the generation of neurotoxic peptide Aβ42. BACE1 is an aspartic protease with four N-glycosylation sites [52]. Site-directed mutagenesis eliminating all BACE1 N-glycans resulted in very slow intracellular maturation suggesting that N-glycans are important in folding and maturation of BACE1 [53]. In the mouse brain, BACE1 contains N-glycans, which are further modified by β1,4-N-acetylglucosaminyltransferase III (GnT-III) that is encoded by the Mgat3 gene [54]. The product of GnT-III is referred to as a bisecting GlcNAc linkage, where GnT-III catalyzes the addition of GlcNAc to a mannose residue linked through a β1,4-linkage [55]. The introduction of the bisecting GlcNAc prevents further processing because this structure cannot act as a substrate for other glycosyltransferases [54]. Post-mortem evaluation of AD patients revealed that the level of bisecting GlcNAc on BACE1 is elevated with disease progression, suggesting that this abnormal change in BACE1 glycosylation may be involved in AD pathogenesis by modulating β-cleavage of APP [54]. It has been well established that oxidative stress plays a crucial role in the neurodegeneration and, more specifically, it induces an up-regulation of BACE1 expression leading to enhance Aβ42 accumulation [56]. Kizuka and colleagues demonstrated that bisecting GlcNAc could modulate the expression of BACE1 induced by oxidative stress. In particular, Kizuka and colleagues demonstrated that the absence of bisecting GlcNAc drives BACE1 towards the lysosomal degradation of BACE1, which, in turn, leads to a reduction in Aβ42 deposition. H2O2-treated Mgat3−/− cells showed a rapid degradation of BACE1 that was abolished by inhibiting lysosomal hydrolases by chloroquine. These results demonstrated that oxidative stress-induced high BACE1 levels were significantly reduced in Mgat3−/− cells due to accelerated lysosomal degradation. GnT-III-deficient (Mgat3−/−) mice, which do not demonstrate obvious gross phenotypic abnormalities, were crossed with APP23 transgenic mouse model of AD, which expresses human APP (hAPP) [57]. Interestingly, in these hAPP/Mgat3−/− mice, the authors found significant reductions of both Aβ40 and Aβ42 levels in an age-dependent manner, which was accompanied by a markedly decreased number of Aβ plaques and an amelioration of the cognitive impairment. These latter results suggest that inhibiting GnT-III may be a promising therapeutic strategy with an advantageous profile of adverse drug reactions compared to BACE1 inhibitors. In this regard, preclinical studies demonstrated that BACE1-deficient mice displayed severe abnormalities including schizophrenia-like behaviors, hypomyelination and early lethality, which suggests that BACE1 inhibitors may exert alarming adverse effects [58][59]. This is further confirmed by the high failure and premature termination rate of recent clinical trials of drugs targeting BACE1, many of which were at an advanced stage [60].
γ–secretase catalyzes the second proteolytic cleavage of APP within its transmembrane region after the proteolytic interventions of α–secretase or BACE1 [52]. γ-secretase is a protease complex and consists of four essential subunits: presenilin (PSEN) (including PSEN1 and 2), nicastrin, anterior pharynx-defective 1 (Aph-1) and presenilin enhancer 2 (Pen-2) [61][62][63][64][65]. γ-secretase is involved in a vast range of crucial biological activities and, besides APP, it cleaves the Notch intracellular domain (NICD), which is able to translocate into the nucleus and regulate gene expression controlling cell proliferation, survival, positioning and differentiation [61][62]. Therefore, when γ-secretase was considered as a potential key target for the development of AD-modifying therapeutics, its genetic and pharmacological manipulation revealed severe developmental defects and serious safety issues [66][67]. Nicastrin is the only glycosylated subunit having 16 possible N-glycosylation sites and the mature protein contains a mixture of high-mannose and complex oligosaccharide side chains generated during trafficking through the Golgi apparatus [68]. PSEN1/2 play a crucial role in the glycosylation process and maturation of nicastrin, as demonstrated in Psen1/2 double-knockout cells, where the mature form of nicastrin is completely lost. Surprisingly, both Aβ and NICD productions were not affected by pharmacological inhibition of mannosidase-I, suggesting that nicastrin glycosylation is not required for γ-secretase activity. However, it is required for the maturation of the protein before its incorporation into the γ-secretase complex or its cell-surface localization . To further understand the relationship between the O-GlcNAcylation of γ-secretase and AD neuropathology, a mouse model of AD carrying 5 × FAD genes was treated with NButGT, a specific OGA inhibitor [69]. NButGT-treated 5 × FAD mice showed reduced levels of Aβ42 and Aβ40 peptides leading to a reduction in amyloid plaque formation, attenuated neuroinflammation and improvements in cognition. Interestingly, NButGT treatment decreased γ-secretase activity without affecting the α- or β-secretase activity . More research will be needed in the near future to better understand the structure-function relationship of γ-secretase in order to identify more specific inhibitors, which selectively modulate Aβ production without affecting Notch cleavage.
Neprilysin is a zinc metallopeptidase identified in the brain and it is the major rate-limiting enzyme involved in Aβ42 degradation [70][71]. Brain regions with high plaque deposition, including the hippocampus and temporal gyrus, were inversely correlated with a selective reduction in neprilysin expression, suggesting that downregulation of neprelysin levels could promote Aβ deposition in the brain [70][71]. The expression, stability and enzymatic activity of neprilysin were affected by N-glycosylation and its activity completely lost upon N-glycosylation removal induced by glycopeptidase F treatment. Furthermore, site-directed mutagenesis of the N-glycosylation sites in neprilysin reveals that an N-glycan at Asn628 is crucial not only for neprilysin expression but also modulates its activity that affects Aβ clearance [70][71][72]. All the above-mentioned mechanisms are depicted in Figure 2.
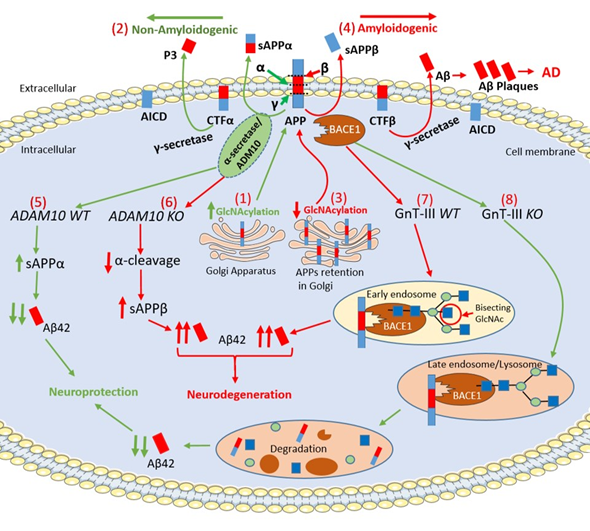
Figure 2. Schematic representation of APP intracellular maturation and its cleavage by secretases. (1) β-Amyloid precursor protein (APP) undergoes glycosylation in the Golgi apparatus and, then, translocates to cell membrane for secretase cleavage (2) The increase of APP glycosylation drives toward the non-amyloidogenic pathway. α-secretase cleaves APP within the β-Amyloid (Aβ) sequence producing the C-terminal fragment α (CTFα) and soluble N-terminal fragment α (sAPPα), which has neuroprotective properties. Afterwards, γ-secretase cleaves CTFα into N-terminal fragment (P3) and a soluble APP intracellular domain (AICD). (3) The pharmacological inhibition of APP glycosylation retains the protein in the Golgi apparatus preventing its transport to cell membrane. The reduction of the glycosylation process drives the amyloidogenic pathway. (4) In the amyloidogenic pathway, β-secretase cleaves APP producing soluble N-terminal fragment β (sAPPβ) and membrane-bound C-terminal fragment β (CTFβ). Then, γ-secretase processes CTFβ into AICD and Aβ42, which forms extracellular Aβ plaques. (5) α-secretase is a member of ‘a disintegrin and metalloprotease’ (ADAM) family and, in particular, ADAM10 cleaves APP increasing the levels of sAPPα leading to neuroprotection. (6) The genetic deletion of ADAM10 suppresses the α-cleavage of APP, decreases in sAPPα levels and increases Aβ42 formation in ADAM10 knockout (KO) mice (Adam10-/-). (7) β-site APP cleaving enzyme-1 (BACE1) contains N-glycans, which are further modified by a glycosyltransferases, namely β1,4-N-acetylglucosaminyltransferase (GnT-III) that is encoded by the Mgat3 gene. In GnT-III WT mice (Mgat3+/+ mice), BACE1 localizes in early endosomes and is modified by GnT-III to bear bisecting GlcNAc generating Aβ42 elevation. (8) In contrast, in GnT-III knockout (KO) mice (Mgat−/− mice), BACE1 is relocated to late endosomes/lysosomes where BACE1 is degraded leading to a decrease of Aβ42 levels. (Red/green up arrows: Increase; Red/green down arrows: Decrease).
4. Conclusions
The present review provides evidence that the alterations of the glycosylation process play an important role in the aetiology and pathogenesis of AD. Despite some conflicting results, preclinical and clinical studies point to an impairment/downregulation of O-GlcNAcylation during the progression of AD. Therefore, this review supports the idea that the glucose metabolic pathway may represent an alternative therapeutic option for targeting AD. In this perspective, the pharmacological modulation of O-GlcNAcylation levels may represent a ‘sweet approach’ to treat AD targeting new mechanisms independent of the amyloid cascade and with comparable impacts in familial and sporadic AD.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21207739
References
- Joiner, C.M.; Li, H.; Jiang, J.; Walker, S. Structural characterization of the O-GlcNAc cycling enzymes: Insights into substrate recognition and catalytic mechanisms. Curr. Opin. Struct. Biol. 2019, 56, 97–106. [Google Scholar] [CrossRef]
- Gao, Y.; Wells, L.; Comer, F.I.; Parker, G.J.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J. Biol. Chem. 2001, 276, 9838–9845. [Google Scholar] [CrossRef]
- Li, S.; Wang, J.; Zang, L.; Zhu, H.; Guo, J.; Zhang, J.; Wen, L.; Chen, Y.; Li, Y.; Chen, X.; et al. Production of glycopeptide derivatives for exploring substrate specificity of human OGA toward sugar moiety. Front. Chem. 2018, 6, 646. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Nam, Y.; Jiang, J.; Sliz, P.; Walker, S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 2011, 469, 564–567. [Google Scholar] [CrossRef]
- Okuyama, R.; Marshall, S. UDP-N-acetylglucosaminyl transferase (OGT) in brain tissue: Temperature sensitivity and subcellular distribution of cytosolic and nuclear enzyme. J. Neurochem. 2003, 86, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Gao, Y.; Mahoney, J.A.; Vosseller, K.; Chen, C.; Rosen, A.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Further characterization of the nucleocytoplasmic β-N-acetylglucosaminidase, O-GlcNAcase. J. Biol. Chem. 2002, 277, 1755–1761. [Google Scholar] [CrossRef]
- Comtesse, N.; Maldener, E.; Meese, E. Identification of a nuclear variant of MGEA5, a cytoplasmic hyaluronidase and a β-N-acetylglucosaminidase. Biochem. Biophys. Res. Commun. 2001, 283, 634–640. [Google Scholar] [CrossRef]
- Keembiyehetty, C.N.; Krzeslak, A.; Love, D.C.; Hanover, J.A. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J. Cell Sci. 2011, 124, 2851–2860. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.Y.; Chatham, J.C.; Darley-Usmar, V.; McMahon, L.L.; Zhang, J. O-GlcNAcylation and neurodegeneration. Brain Res. Bull. 2017, 133, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.; Xu, M.; Davey, A.K.; Danon, J.J.; Mellick, G.D.; Kassiou, M.; Rudrawar, S. O-GlcNAc modification protects against protein misfolding and aggregation in neurodegenerative disease. ACS Chem. Neurosci. 2019, 10, 2209–2221. [Google Scholar] [CrossRef]
- Olivier-Van Stichelen, S.; Wang, P.; Comly, M.; Love, D.C.; Hanover, J.A. Nutrient-driven O-linked N-acetylglucosamine (O-GlcNAc) cycling impacts neurodevelopmental timing and metabolism. J. Biol. Chem. 2017, 292, 6076–6085. [Google Scholar] [CrossRef]
- Pinho, T.S.; Verde, D.M.; Correia, S.C.; Cardoso, S.M.; Moreira, P.I. O-GlcNAcylation and neuronal energy status: Implications for Alzheimer’s disease. Ageing Res. Rev. 2018, 46, 32–41. [Google Scholar] [CrossRef]
- Clark, R.J.; McDonough, P.M.; Swanson, E.; Trost, S.U.; Suzuki, M.; Fukuda, M.; Dillmann, W.H. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J. Biol. Chem. 2003, 278, 44230–44237. [Google Scholar] [CrossRef]
- Li, X.; Lu, F.; Wang, J.Z.; Gong, C.X. Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur. J. Neurosci. 2006, 23, 2078–2086. [Google Scholar] [CrossRef]
- Lim, S.; Haque, M.M.; Nam, G.; Ryoo, N.; Rhim, H.; Kim, Y.K. Monitoring of intracellular tau aggregation regulated by OGA/OGT inhibitors. Int. J. Mol. Sci. 2015, 16, 20212–20224. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, K.T.; Iverfeldt, K. O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-β precursor protein (APP). Biochem. Biophys. Res. Commun. 2011, 404, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, G.E.; Macauley, M.S.; Stubbs, K.A.; Dennis, R.J.; Taylor, E.J.; Davies, G.J.; Greig, I.R.; Vocadlo, D.J. Analysis of PUGNAc and NAG-thiazoline as transition state analogues for human O-GlcNAcase: Mechanistic and structural insights into inhibitor selectivity and transition state poise. J. Am. Chem. Soc. 2007, 129, 635–644. [Google Scholar] [CrossRef]
- Wang, Z.; Gucek, M.; Hart, G.W. Cross-talk between GlcNAcylation and phosphorylation: Site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. USA 2008, 105, 13793–13798. [Google Scholar] [CrossRef] [PubMed]
- Yuzwa, S.A.; Shan, X.; Jones, B.A.; Zhao, G.; Woodward, M.L.; Li, X.; Zhu, Y.; McEachern, E.J.; Silverman, M.A.; Watson, N.V.; et al. Pharmacological inhibition of O-GlcNAcase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/APP mutant mice. Mol. Neurodegener. 2014, 9, 42. [Google Scholar] [CrossRef]
- Macauley, M.S.; Shan, X.; Yuzwa, S.A.; Gloster, T.M.; Vocadlo, D.J. Elevation of global O-GlcNAc in rodents using a selective O-GlcNAcase inhibitor does not cause insulin resistance or perturb glucohomeostasis. Chem. Biol. 2010, 17, 949–958. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; He, Y.; Whitworth, G.E.; Stubbs, K.A.; McEachern, E.J.; Davies, G.J.; et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008, 4, 483–490. [Google Scholar] [CrossRef]
- Paul, S.; Haskali, M.B.; Liow, J.S.; Zoghbi, S.S.; Barth, V.N.; Kolodrubetz, M.C.; Bond, M.R.; Morse, C.L.; Gladding, R.L.; Frankland, M.P.; et al. Evaluation of a PET radioligand to image O-GlcNAcase in brain and periphery of rhesus monkey and knock-out mouse. J. Nucl. Med. 2019, 60, 129–134. [Google Scholar] [CrossRef]
- Videira, P.A.Q.; Castro-Caldas, M. Linking glycation and glycosylation with inflammation and mitochondrial dysfunction in Parkinson’s disease. Front. Neurosci. 2018, 12, 381. [Google Scholar] [CrossRef] [PubMed]
- Gallelli, C.A.; Calcagnini, S.; Romano, A.; Koczwara, J.B.; de Ceglia, M.; Dante, D.; Villani, R.; Giudetti, A.M.; Cassano, T.; Gaetani, S. Modulation of the oxidative stress and lipid peroxidation by endocannabinoids and their lipid analogues. Antioxidants 2018, 7, 93. [Google Scholar] [CrossRef]
- Trabace, L.; Cassano, T.; Tucci, P.; Steardo, L.; Kendrick, K.M.; Cuomo, V. The effects of nitric oxide on striatal serotoninergic transmission involve multiple targets: An in vivo microdialysis study in the awake rat. Brain Res. 2004, 1008, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Griffith, C.M.; Macklin, L.N.; Cai, Y.; Sharp, A.A.; Yan, X.X.; Reagan, L.P.; Strader, A.D.; Rose, G.M.; Patrylo, P.R. Impaired glucose tolerance and reduced plasma insulin precede decreased AKT phosphorylation and GLUT3 translocation in the hippocampus of old 3 − Tg-AD Mice. J. Alzheimer Dis. 2019, 68, 809–837. [Google Scholar] [CrossRef]
- Macklin, L.; Griffith, C.M.; Cai, Y.; Rose, G.M.; Yan, X.X.; Patrylo, P.R. Glucose tolerance and insulin sensitivity are impaired in APP/PS1 transgenic mice prior to amyloid plaque pathogenesis and cognitive decline. Exp. Gerontol. 2017, 88, 9–18. [Google Scholar] [CrossRef]
- Pardeshi, R.; Bolshette, N.; Gadhave, K.; Ahire, A.; Ahmed, S.; Cassano, T.; Gupta, V.B.; Lahkar, M. Insulin signaling: An opportunistic target to minify the risk of Alzheimer’s disease. Psychoneuroendocrinology 2017, 83, 159–171. [Google Scholar] [CrossRef]
- Cisternas, P.; Salazar, P.; Silva-Alvarez, C.; Barros, L.F.; Inestrosa, N.C. Activation of Wnt signaling in cortical neurons enhances glucose utilization through glycolysis. J. Biol. Chem. 2016, 291, 25950–25964. [Google Scholar] [CrossRef]
- Cisternas, P.; Zolezzi, J.M.; Martinez, M.; Torres, V.I.; Wong, G.W.; Inestrosa, N.C. Wnt-induced activation of glucose metabolism mediates the in vivo neuroprotective roles of Wnt signaling in Alzheimer disease. J. Neurochem. 2019, 149, 54–72. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, L.R.; Spencer, Z.T.; Nayak, A.; Cselenyi, C.S.; Benchabane, H.; Youngblood, C.Q.; Zouaoui, A.; Ng, V.; Stephens, L.; Hann, T.; et al. Developmental regulation of Wnt signaling by Nagk and the UDP-GlcNAc salvage pathway. Mech. Dev. 2019, 156, 20–31. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, M.; Sun, J.; Guo, A.; Fernando, R.L.; Chen, Y.; Peng, P.; Zhao, G.; Deng, Y. DPP-4 inhibitor improves learning and memory deficits and AD-like neurodegeneration by modulating the GLP-1 signaling. Neuropharmacology 2019, 157, 107668. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin reductase-a mediates the beneficial effects of intranasal insulin in Alzheimer disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef] [PubMed]
- Peila, R.; Rodriguez, B.L.; Launer, L.J.; Honolulu-Asia aging, S. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia aging study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef]
- Akasaka-Manya, K.; Manya, H.; Sakurai, Y.; Wojczyk, B.S.; Spitalnik, S.L.; Endo, T. Increased bisecting and core-fucosylated N-glycans on mutant human amyloid precursor proteins. Glycoconj. J. 2008, 25, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Kirino, Y.; Suzuki, T. Cleavage of Alzheimer’s amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway. Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J. Biol. Chem. 1998, 273, 6277–6284. [Google Scholar] [CrossRef]
- McFarlane, I.; Georgopoulou, N.; Coughlan, C.M.; Gillian, A.M.; Breen, K.C. The role of the protein glycosylation state in the control of cellular transport of the amyloid β precursor protein. Neuroscience 1999, 90, 15–25. [Google Scholar] [CrossRef]
- Nakagawa, K.; Kitazume, S.; Oka, R.; Maruyama, K.; Saido, T.C.; Sato, Y.; Endo, T.; Hashimoto, Y. Sialylation enhances the secretion of neurotoxic amyloid-beta peptides. J. Neurochem. 2006, 96, 924–933. [Google Scholar] [CrossRef]
- Chun, Y.S.; Kwon, O.H.; Chung, S. O-GlcNAcylation of amyloid-β precursor protein at threonine 576 residue regulates trafficking and processing. Biochem. Biophys. Res. Commun. 2017, 490, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Cheng, B.; Culwell, A.R.; Esch, F.S.; Lieberburg, I.; Rydel, R.E. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron 1993, 10, 243–254. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F. α-secretase in Alzheimer’s disease: Molecular identity, regulation and therapeutic potential. J. Neurochem. 2011, 116, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [PubMed]
- Escrevente, C.; Morais, V.A.; Keller, S.; Soares, C.M.; Altevogt, P.; Costa, J. Functional role of N-glycosylation from ADAM10 in processing, localization and activity of the enzyme. Biochim. Biophys. Acta 2008, 1780, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Kizuka, Y.; Nakano, M.; Kitazume, S.; Saito, T.; Saido, T.C.; Taniguchi, N. Bisecting GlcNAc modification stabilizes BACE1 protein under oxidative stress conditions. Biochem. J. 2016, 473, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Vanoni, O.; Paganetti, P.; Molinari, M. Consequences of individual N-glycan deletions and of proteasomal inhibition on secretion of active BACE. Mol. Biol. Cell 2008, 19, 4086–4098. [Google Scholar] [CrossRef] [PubMed]
- Kizuka, Y.; Kitazume, S.; Fujinawa, R.; Saito, T.; Iwata, N.; Saido, T.C.; Nakano, M.; Yamaguchi, Y.; Hashimoto, Y.; Staufenbiel, M.; et al. An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease. EMBO Mol. Med. 2015, 7, 175–189. [Google Scholar] [CrossRef]
- Stanley, P. Biological consequences of overexpressing or eliminating N-acetylglucosaminyltransferase-TIII in the mouse. Biochim. Biophys. Acta 2002, 1573, 363–368. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar] [CrossRef]
- Priatel, J.J.; Sarkar, M.; Schachter, H.; Marth, J.D. Isolation, characterization and inactivation of the mouse Mgat3 gene: The bisecting N-acetylglucosamine in asparagine-linked oligosaccharides appears dispensable for viability and reproduction. Glycobiology 1997, 7, 45–56. [Google Scholar] [CrossRef]
- Savonenko, A.V.; Melnikova, T.; Laird, F.M.; Stewart, K.A.; Price, D.L.; Wong, P.C. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc. Natl. Acad. Sci. USA 2008, 105, 5585–5590. [Google Scholar] [CrossRef]
- Willem, M.; Garratt, A.N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C. Control of peripheral nerve myelination by the β-secretase BACE1. Science 2006, 314, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Francis, R.; McGrath, G.; Zhang, J.; Ruddy, D.A.; Sym, M.; Apfeld, J.; Nicoll, M.; Maxwell, M.; Hai, B.; Ellis, M.C.; et al. aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of βAPP, and presenilin protein accumulation. Dev. Cell 2002, 3, 85–97. [Google Scholar] [CrossRef]
- Goutte, C.; Tsunozaki, M.; Hale, V.A.; Priess, J.R. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. USA 2002, 99, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Winkler, E.; Edbauer, D.; Prokop, S.; Basset, G.; Yamasaki, A.; Kostka, M.; Haass, C. PEN-2 is an integral component of the γ-secretase complex required for coordinated expression of presenilin and nicastrin. J. Biol. Chem. 2002, 277, 39062–39065. [Google Scholar] [CrossRef]
- Yu, G.; Nishimura, M.; Arawaka, S.; Levitan, D.; Zhang, L.; Tandon, A.; Song, Y.Q.; Rogaeva, E.; Chen, F.; Kawarai, T.; et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 2000, 407, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Nadeau, P.; Song, W.; Donoviel, D.; Yuan, M.; Bernstein, A.; Yankner, B.A. Presenilins are required for γ-secretase cleavage of β-APP and transmembrane cleavage of Notch-1. Nat. Cell Biol. 2000, 2, 463–465. [Google Scholar] [CrossRef]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907. [Google Scholar] [CrossRef]
- Herreman, A.; Van Gassen, G.; Bentahir, M.; Nyabi, O.; Craessaerts, K.; Mueller, U.; Annaert, W.; De Strooper, B. gamma-Secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J. Cell Sci. 2003, 116, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Nam, D.W.; Park, S.Y.; Song, H.; Hong, H.S.; Boo, J.H.; Jung, E.S.; Kim, Y.; Baek, J.Y.; Kim, K.S.; et al. O-linked β-N-acetylglucosaminidase inhibitor attenuates beta-amyloid plaque and rescues memory impairment. Neurobiol. Aging 2013, 34, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Shirotani, K.; Lu, B.; Gerard, N.P.; Gerard, C.; Hama, E.; Lee, H.J.; Saido, T.C. Metabolic regulation of brain Abeta by neprilysin. Science 2001, 292, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Aβ1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef]
- Sato, B.; Katagiri, Y.U.; Iijima, K.; Yamada, H.; Ito, S.; Kawasaki, N.; Okita, H.; Fujimoto, J.; Kiyokawa, N. The human CD10 lacking an N-glycan at Asn(628) is deficient in surface expression and neutral endopeptidase activity. Biochim. Biophys. Acta 2012, 1820, 1715–1723. [Google Scholar] [CrossRef]