Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Respiratory System
Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive fibrosing interstitial lung disease that occurs predominantly in the older population. It is the most common cause of idiopathic interstitial pneumonias and was previously thought to be a rare disease, however, there is an increasing trend in incidence and prevalence rate globally.
- idiopathic pulmonary fibrosis (IPF)
- treatment
- pharmacotherapy
1. Pathophysiology and Etiology
The pathophysiology of idiopathic pulmonary fibrosis (IPF) involves the destruction of normal lung architecture along with inflammation and fibrosis. While the role of fibrosis is established in the pathogenesis of IPF, the role of inflammation remains controversial given multiple failed anti-inflammatory therapies as IPF treatment [4]. However, it has been hypothesized that chronic inflammation as a result of repeated injury to the lung epithelium partly due to the deposition of protein of the extracellular matrix (ECM) and dysregulated repair responses lead to eventual fibrosis [4,5].
More recently, cytokine transforming growth factor-β (TGF-β) has been implicated in the development of pulmonary fibrosis [5,6]. TGF-β is pivotal in many cellular functions such as proliferation, differentiation, ECM synthesis, and apoptosis [7]. In the human lung, TGF-β is expressed by alveolar epithelial cells (AEC), alveolar macrophages, and fibroblasts, and is known to be elevated in cases of acute lung injuries, making it a critical mediator of inflammation [7,8,9,10]. Crucially, TGF-β induces the differentiation of myofibroblasts from fibroblasts, leading to the production of ECM such as collagen, laminin, and fibronectin [11]. Excessive ECM surrounding inflamed or injured lung tissue inhibits normal physiological functions of the cell including cell repair, and in cases of chronic inflammation, the balance in the production and degradation of ECM is affected due to increased TGF-β, consequently leading to ECM accumulation [12]. Subsequently, this leads to lung tissue remodeling and fibrosis. Understanding the pathophysiology of IPF is critical in the development of novel anti-fibrotics [See Figure 1].
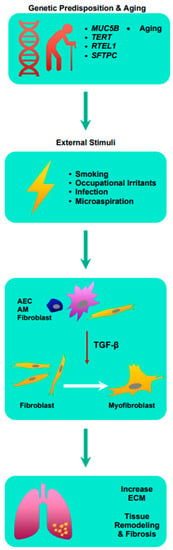
Figure 1. Summary of the pathophysiology of idiopathic pulmonary fibrosis. AEC: airway epithelial cell; AM: alveolar macrophages; ECM: extracellular matrix; MUC5B: Mucin 5B; RTEL1: regulator of telomere length 1; SFTPC: surfactant protein C; TERT: telomerase reverse transcriptase; TGF-β: transforming growth factor-β.
Although the cause of IPF is largely unknown, significant milestones have been achieved in recent years with regard to the etiology of IPF. There are several genetic mutations and external factors that have now been identified that increases the risk of developing IPF.
1.1. Role of Genetic Mutation
IPF, which occur in clusters in families, are referred to as familial interstitial pneumonia (FIP), a phenomenon that has been observed as early as 1950 [13]. There are now several genetic mutations that have been identified that play an important role in the development of IPF. A common polymorphism in the promoter of Mucin 5B, encoded by the gene MUC5B, is associated with both FIP and sporadic pulmonary fibrosis [14]. An intercontinental genetic study found that mutation in MUC5B is the strongest risk variant for IPF; higher with two copies of risk allele vs. one copy of risk allele with an odds ratio of 18.7 and 5.5, respectively [15]. Mucin 5B is a gel forming mucin that is essential in mucociliary clearance and is overexpressed in IPF lungs, leading to mucociliary dysfunction and increased fibrosis [16].
Short telomere syndrome, as a result of telomerase mutations, is another risk factor for FIP. It has been shown in in vitro studies that in the lung of an IPF patient with known telomerase mutation, shorter telomerases are seen in the alveolar epithelium compared to normal individuals [17]. Short telomeres reduce the tissue renewal capacity and telomeres shorten as an individual ages, which means that this could be the reason why the diagnosis of IPF has an age-related onset [17]. Moreover, cell senescence plays an essential role in resisting environmental stress and repopulating injured bronchoalveolar epithelia [15]. A very recent study involving over 120 centers globally studied 2180 cases of IPF using whole-genome sequencing to investigate the role of rare variants on IPF risk found that rare variants within the telomerase reverse transcriptase (TERT) and regulator of telomere length 1 (RTEL1) genes were significantly associated with IPF [18].
Genes encoding for surfactant protein C (SF-C) such as SFTPC have also been implicated in the diagnosis of FIP where a genetic study demonstrated, in a family of five generations, that individuals who were heterozygous for the mutation had biopsy changes consistent with usual interstitial pneumonitis (UIP) or non-specific interstitial pneumonitis (NSIP) [19]. Furthermore, SFTPC mutations have also been identified in sporadic cases of IPF [20]. In a normal mature lung, the pulmonary surfactant is secreted by type 2 AEC and functions by reducing surface tension in the alveolus and stabilizing the alveoli, maintaining lung volumes at end-expiration [21]. Mutations in SFTPC cause abnormal surfactant processing, leading to endoplasmic reticulum stress in type 2 AEC and eventual fibrosis of the lung tissue [22,23].
1.2. Role of Environmental Factors
It has long been speculated that environmental factors or irritants such as cigarette smoke and occupational dust exposure could be a risk factor for developing IPF. Cigarette smoking has been the most studied of them all given its reputation in other chronic lung diseases. Experts believe that cigarette smoking may promote the transformation of lung fibroblasts to myoblasts, contribute to the shortening of the telomeres, and inducing endoplasmic reticulum stress [23,24,25]. A prospective study looking at over 800 individual IPF cases in the UK concluded that both active and maternal tobacco smoking increased the risk of IPF on top of having a synergistic effect with one another [26]. Furthermore, environmental factors may play an additive role to genetic factors contributing to the pathogenicity of IPF, where genetically susceptible lung tissue segments may experience repeated microscopic injuries from environmental irritants, eventually homogenizing into widespread fibrosis [15].
1.3. Role of Infection
Some evidence suggests that the chronic infection of viruses such as EBV, CMV, HHV-7, and HHV-8 increases the risk of developing IPF [27,28]. The basis of this is immunosenescence, where persistent viral infection is thought to cause exhaustion of the immune system overtime, leading to T cell aging [29]. As aging is associated with IPF, some experts believe that immunosenescence is a risk factor for developing IPF [30]. More recently, SARS-Co-V2 infections was observed to increase pro-fibrotic macrophages, similar to those seen in IPF patients [31]. While at present the role of bacterial infection in the induction of IPF is unclear, it is thought to be more likely linked to the progression rather than initiation of fibrosis in patients [32,33].
1.4. Aging and Cellular Senescence
While genetic pre-disposition (i.e., mutations in telomeres) may lead to premature aging, physiological aging itself is a risk factor for developing IPF. Cellular senescence is a form of irreversible permanent cell cycle arrest and occurs naturally with aging, however, it may occur at any stage of an individual’s life [34]. The quantity of senescent cells increases with age as well as in aging-related diseases such as IPF, but the underlying mechanism that leads to physiological or pathological cellular senescence remains to be elucidated [35]. Senescence from normal aging on top of induced senescence from smoking, occupational irritants, or infection may accelerate cellular senescence. The implications of accelerated senescence are dysregulations of the innate and adaptive immune system including abnormal AEC and fibroblast activation as well as increased oxidative stress leading to impaired cell repair [36].
2. Diagnosis
The diagnosis of IPF requires clinical co-relation with the presence of a typical pattern of usual interstitial pneumonia (UIP), identified either histologically or via computed topography [37]. Histopathological hallmarks of UIP are a combination of four features that are: (1) patchy dense fibrosis with architectural distortion; (2) predilection for subpleural or paraseptal lung parenchymal; (3) presence of fibroblastic foci; and (4) absence of features of alternative diagnoses [2]. Radiological features of UIP include the presence of honey combing with or without traction bronchiectasis, ground glass opacification, and interlobular septal thickening along with typical distribution in the subpleural or bases [2]. Based on the radiological and or histological features, the diagnosis of IPF is described as definite, likely, indeterminate, or alternate diagnosis suspected [2].
This entry is adapted from the peer-reviewed paper 10.3390/life13020486
This entry is offline, you can click here to edit this entry!