Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Orthopedics
The epithelial to mesenchymal transition (EMT) is a process by which cells exhibiting an epithelial phenotype adopt a mesenchymal phenotype, which facilitates migration, invasion, and metastasis. This article provides an overview of the role, regulation, and targeting of EMT in Osteosarcoma, a primary bone malignancy affecting mainly children and young adults.
- osteosarcoma
- epithelial-mesenchymal transition
- transcriptional regulation
1. Introduction
Osteosarcoma (OS) is a primary bone malignancy with an annual incidence of 2–4 per million [1]. It typically affects children, teens, and young adults [2], with a peak incidence from ages 10–19 [1], a second peak in adults over 60 [2], and a slight male preponderance [3]. The overall 5-year survival rate for OS is 60% but decreases to 27% in the presence of distant metastases [4]; the rate of metastases at diagnosis is 18% [5].
The origin of OS is poorly understood. As a sarcoma, it arises from mesenchymal cells, but it is not currently known whether the precursor cells are osteoblasts or mesenchymal stem cells [6]. Although the etiology of OS is largely a mystery, multiple risk factors have been identified. These include medical conditions such as hereditary retinoblastoma, Li-Fraumeni syndrome, Werner syndrome, Rothmund-Thompson syndrome, Bloom syndrome, and Paget’s disease [3]. Other risk factors include exposure to ionizing radiation and alkylating agents, both of which may have been used in the treatment of a prior malignancy [3].
The mainstay of treatment for osteosarcoma is surgical resection and frequently involves both neoadjuvant and adjuvant chemotherapy for higher grade tumors [7]. While advances in surgical techniques and chemotherapeutic regimens were associated with an initial improvement in outcomes, overall survival in OS has not significantly changed in several decades [8]. As medicine becomes more personalized, there is a growing interest in the identification of novel targeted therapies. A key component in developing targeted therapy is identifying specific pathways, proteins, or other molecules essential to cancer cell function. One of the cellular features often associated with aggressive cancers is the epithelial to mesenchymal transition (EMT).
2. EMT in Cancer
EMT is depicted in Figure 1. It is a process by which cells exhibiting an epithelial phenotype adopt a mesenchymal phenotype, which facilitates migration, invasion, and metastasis [9]. It exists in equilibrium with a reverse and complementary process, the mesenchymal to epithelial transition (MET), wherein cells revert back to an epithelial phenotype. Primary epithelial tumors exhibit epithelial cell markers such as E-cadherin. These cells demonstrate apical polarity, adhesion to a basement membrane, and tight cellular junctions [10]. For many cancers, EMT is critical in the early transition from normal to malignant cells. It is characterized by downregulation of epithelial cell markers, destabilization and loss of cell–cell junctions, loss of adherence to basement membrane and apical polarity, and cytoskeletal reorganization [9]. The result of these changes is a cell with mesenchymal morphology and characteristics.
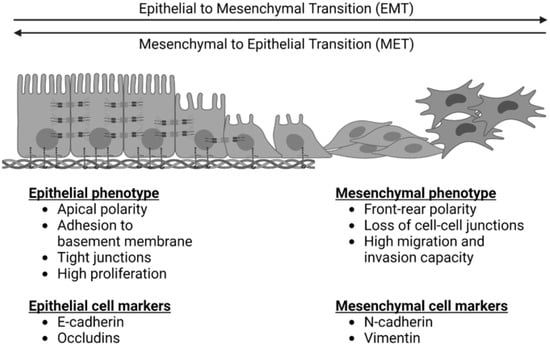
Figure 1. The epithelial to mesenchymal transition (EMT) and the reverse process of the mesenchymal to epithelial transition (MET). EMT is characterized by a loss of epithelial cell markers, an increase in mesenchymal cell markers, a loss of apical cell polarity, a loss of tight cell junctions, and an increased capacity for cell migration and invasion. MET is characterized by a loss of mesenchymal cell markers, an increase in epithelial cell markers, increased apical cell polarity, tight junctions, adherence to a basement membrane, and increased cell proliferation.
Given the migratory potential of mesenchymal cells compared to epithelial cells, EMT has long been linked to cancer metastasis. However, inhibition of EMT has not been shown to affect the establishment of cancer metastases in vivo [11,12], and the cells found within metastatic tumors are more likely to exhibit an epithelial phenotype [12,13]. Despite this, tumor cells that have undergone EMT appear to drive local invasion and angiogenesis of the primary tumor [13]. These results suggest that EMT is critical for tumor invasion into the local vascular system, allowing cells to migrate to distant organs where secondary tumors are established largely by cells with an epithelial phenotype, which have a greater propensity for proliferation [9]. These may be either cells that have undergone EMT and subsequently MET or primary tumor cells that did not undergo EMT [13].
The molecular pathways associated with EMT are summarized in Figure 2. Zinc-finger E-box binding homeobox (ZEB), snail family transcriptional repressor 1 (SNAIL), snail family transcriptional repressor 2 (SLUG), and twist-related protein (TWIST) are well-known EMT transcription factors that are established downstream targets of multiple signaling pathways, including the canonical wnt/β-catenin pathway, the neurogenic locus notch homolog protein (Notch) pathway, the Transforming Growth Factor β/Suppressor of Mothers Against Decapentaplegic (TGFβ/SMAD) pathway, the phosphoinositide 3-kinase (PI3K)/Akt pathway, the mitogen-activated protein kinase (MAPK) pathway, the Ras/Raf/Mitogen-activated protein kinase/ERK kinase/extracellular-signal-regulated kinase (RAS/RAF/MEK/ERK) axis, and the Janus kinase-Signal Transducer and Activator of Transcription JAK/STAT pathway [10]. These signaling cascades often interact, share many intermediaries, and impact the regulation of one another. This presents a challenge for studying and targeting EMT, as the individual pathways are difficult to isolate.
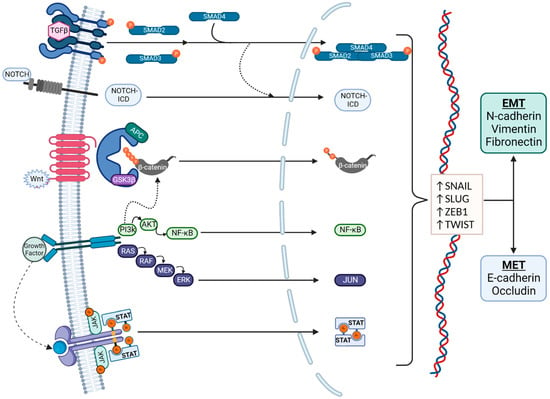
Figure 2. Signaling pathways in EMT. EMT regulation is complex and affected by multiple pathways, which also interact with each other. Regulation is typically via the Transforming Growth Factor β (TGFβ)/SMAD, Notch, canonical wnt, phosphoinositide 3-kinase (PI3K)/Akt, RAS/RAF, and JAK/STAT pathways. The transcription factors that mediate EMT are primarily Snail, Slug, ZEB1, and TWIST. EMT is characterized by an increased production of N-cadherin, vimentin, and fibronectin, and MET is characterized by an increased production of E-cadherin and Occludin.
3. EMT Signaling Pathways
3.1. TGFβ/SMAD Pathway
The TGFβ family of proteins includes three TGFβ isoforms, activins, and bone morphogenic proteins (BMPs) [14]. In EMT, TGFβs bind to TGFβ receptors (1/2), which initiate a signaling cascade, leading to the increased transcription of genes involved in EMT. Binding of TGFβ to its receptors (1/2) leads to phosphorylation of SMAD2 and SMAD3, which then form a complex with SMAD4. BMPs also bind TGFβ receptors, activating SMAD1 and SMAD5 and then forming a complex with SMAD4. These trimeric complexes migrate to the nucleus to act as transcription factors.
SMAD complexes activate the mesenchymal genes vimentin and fibronectin, as well as the EMT transcription factors Snail, Slug, Zinc finger E-box-binding homeobox 1 (ZEB1) and Twist. These, in turn, repress E-cadherin and can upregulate the expression of TGFβ ligands, establishing a positive feedback loop to maintain EMT [9,15,16].
3.2. Canonical wnt Pathway
The canonical wnt pathway is considered to be a key activator of EMT [9]. Signaling is initiated by a group of wnt ligands that bind to Frizzled receptors and trigger a cascade of events, leading to the nuclear translocation of β-catenin. β-catenin is constitutively produced in the cell and stored in cytosolic pools. In the absence of wnt signaling, phosphorylated β-catenin is associated with a destruction complex, ubiquitinated, and degraded by proteasomes. Following activation of the canonical wnt pathway, β-catenin is dephosphorylated and translocates to the nucleus, where it acts as a transcriptional co-factor to induce the expression of genes involved in cell differentiation, proliferation, and tumorigenesis [17,18].
This pathway has been directly implicated in EMT via the expression of Twist, Slug, N-cadherin, and the repression of E-cadherin [19]. The known EMT transcription factor Snail has been shown to positively regulate wnt signaling [20]. The inhibition of Secreted Frizzled Related Protein 1 (SFRP), a negative regulator of wnt ligands, has also been shown to have EMT-like effects in breast carcinoma cells in vitro, while sensitizing them to TGFβ-induced EMT [21].
β-catenin is also located at the cell membrane as part of an E-cadherin-containing multi-component adherens junction complex, which is a component of cell–cell interaction junctions. β-catenin contributes to anchoring E-cadherin, a transmembrane cell–cell adhesion protein at the cell surface to the intracellular actin cytoskeleton. β-catenin is released from the adherens complex upon disruption of these adherens junctions between cells. Once available in the cytosol, it enters the pathway described above and is either phosphorylated and degraded or, if the wnt pathway is active, dephosphorylated and translocated to the nucleus to function as a transcription factor for EMT-genes [22]. E-cadherin can therefore act as a negative regulator of the canonical wnt pathway by sequestering most of the β-catenin in the epithelial cell membrane.
3.3. Notch Pathway
The Notch pathway has been implicated in inducing EMT in both normal and neoplastic tissues, and is involved in controlling cell fate, differentiation, and proliferation. Four isoforms (Notch1 through Notch4) are known to bind Delta-like or Jagged family ligands. This interaction triggers a series of proteolytic events leading to the active fragment Notch Intracellular Domain (Notch ICD), which then acts in the nucleus, where it associates with binding partners and transcriptional activators [23]. Several components of the Notch pathway are highly expressed at the invasive margins of tumors, which express EMT markers such as vimentin, suggesting an important role for the Notch pathway in the regulation of EMT [24]. Notch acts via transcriptional regulation of ZEB, Snail, and Slug, which repress expression of E-cadherin and induce expression of vimentin and fibronectin [23,24,25].
There is crosstalk between the Notch and TGFβ pathway that occurs via SMADs. As described above, the SMAD family of proteins are integral to TGFβ signaling. They have also been shown to associate with Notch-ICD. This affects the expression of genes downstream of both Notch and TGFβ that are required for mesenchymal differentiation, a key component in EMT [26]. Silencing components of the Notch pathway have also been shown to prevent TGFβ-induced EMT [27].
3.4. Tyrosine Kinase Pathways
Mitogenic growth factors also play a role in the regulation of EMT. The binding of these growth factors causes their receptors to dimerize and induces the activation of both receptor and non-receptor tyrosine kinases (TKs). This enables the activation of several pathways—including the MAPK, JAK-STAT, and phosphatidylinositol 3-kinase-Akt (PI3-Akt) pathways. All of these have been implicated in EMT, and are involved in cell growth, proliferation, and migration [28]. PI3K/Akt has also been shown to play an important role in the regulation of transcriptionally active β-catenin, a key molecule in the previously discussed wnt signaling pathway [29]. Inhibition of TKs is a growing field of study in cancer therapeutics, with multiple inhibitors currently under investigation [30].
Inhibition of fibroblast growth factor (FGF), a mitogenic growth factor that participates in the induction of EMT via activation of the MAPK, induces the reverse process MET in vitro and delays tumor growth in vivo [31]. One isoform, FGF2, has been associated with reduced overall survival in several carcinoma types if overexpressed [31].
The binding of epidermal growth factor (EGF) to its receptor leads to activation of MAPK pathway and decreased expression of E-cadherin [32]. EGF also activates the JAK2-STAT3 pathway, which leads to EMT activation via Twist [33]. Additionally, EGF has been shown to induce EMT via TGFβ signaling and regulation of Snail [34] and phosphorylation of SMAD2/SMAD3 [35].
The activation of Akt, or Protein Kinase B, has been shown to upregulate the phosphorylation of Twist1 and inhibit apoptosis [36], and the inhibition of Akt has been shown to induce MET [37]. For example, hepatocyte growth factor (HGF) has been shown to activate EMT [38], which can be reversed via inhibition of the PI3K/Akt pathway. HGF was found to enhance tumor progression and metastasis of hepatocellular carcinoma in association with the c-MET receptor tyrosine kinase [39], a known activator of PI3K/Akt.
4. EMT in OS
As a mesenchymal cancer, the importance of EMT in OS has been disputed [40,41]. In fact, an early investigation including 4 clinical osteosarcoma samples by Sato et al. found there was no detectable E-cadherin expression in these cells [42], suggesting that the repression/downregulation of E-cadherin—a classically described step in EMT—would not be possible. In contrast, Yin et al. found that 20.6% of OS tissue samples expressed E-cadherin and those that did were less likely to metastasize, whereas the expression of Twist was significantly related to metastases and poorer overall survival [43]. The promotion of EMT in OS characterized by increased migration and invasion in vitro has been shown to be mediated via upregulation of Snail [44,45,46,47,48,49,50,51,52,53,54,55,56,57], Slug [58,59], Twist [60,61,62,63], and ZEB [64,65,66,67].
The following sections give an overview of studies that have examined the roles of different EMT regulatory molecules in OS. All of these were found to affect the expression of EMT-related cell-markers and are correlated closely with EMT-associated cellular features such as increased migration and invasion. Many also showed a link between their proposed EMT-regulatory molecule and OS metastases in vivo in animal models. Taken together, these results suggest that EMT does play a role in osteosarcoma and is associated with a more aggressive tumor phenotype. However, the term “transition” is not ideally suited to sarcoma cancers, and EMT may be better thought of as a set of pathways utilized to maintain and promote the existing mesenchymal phenotype.
Sannino et al. posited a possible hybrid phenotype in sarcoma tumors cells, utilizing the EMT and MET pathways to acquire both mesenchymal and epithelial characteristics that favor the initiation and establishment of distant metastases [40].
The highlighted pathways important in EMT regulation have all been shown to have a role in OS. TGFβs promote EMT and metastases in OS [68], and TGFβ inhibition has been shown to decrease EMT in OS [58,69,70,71,72,73,74]. Chen et al. also identified that estrogen-related receptor α (ERRα) upregulates TGF-β-mediated EMT in two OS cell lines [50]. Others have highlighted roles for MAPK [63,75,76,77] and JAK/STAT [52,78,79,80,81,82].
Notch signaling promotes proliferation, migration, and invasion of OS cells, and Notch overexpression increased tumor growth in vivo [83,84,85,86]. Notch inhibition reduced chemo-resistance in OS in vitro [87,88]. Wnt signaling has also been shown to mediate EMT in OS [49,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104]. It has been proposed that wnt signaling is particularly important in the pathogenesis of OS cancer stem cells [105].
TKs are of particular interest in OS, and multiple different TK proteins have been associated with aggressive cellular phenotypes. Many studies have demonstrated a regulatory role in OS for the downstream TK pathways PI3K/Akt [106,107,108,109,110,111,112,113,114] and RAS/RAF/MEK/ERK [61,115,116,117,118]. Multiple TK inhibitors have been a part of recently completed or ongoing clinical trials in the treatment of OS, including Apatinib, Axitinib, Cabozantinib, Cediranib, Crizotinib, Dasatinib, Imatinib, Pazopanib, Regorafenib, Sorafenib, and Sunitinib [119,120].
5. Regulation of EMT in OS—Proteins
As a complex and multi-faceted process, several proteins have been implicated in EMT regulation in OS [44,48,49,53,54,55,61,62,64,78,89,91,93,94,97,100,106,107,108,109,112,113,114,116,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151]. These proteins were found to either promote [44,48,53,54,61,64,78,89,93,94,97,100,106,107,108,109,112,113,114,116,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146] or inhibit [49,55,62,91,147,148,149,150,151] EMT in vitro, and the majority were found to be correspondingly upregulated or downregulated in clinical OS tissue samples and/or established cell lines compared to normal controls. Each group of authors found a significant correlation between the studied protein and the levels of EMT-related proteins, such as E-cadherin, N-cadherin, and vimentin. They also reported a significant effect on aggressive cellular characteristics, such as migration and invasion ability in vitro. Where noted, the results were confirmed in vivo with mouse xenograft experiments.
A detailed review of the individual proteins investigated for their regulatory role in EMT of OS cells is outside of the scope of this review. Generally, their endogenous functions can be grouped into the following families: cell cycle regulation, immunity/inflammatory, cell signaling, cell structure, and metabolism. Each of these categories has a logical impact on EMT and/or cancer cell behavior.
Changes to cell differentiation and cell cycle regulation are recognized mechanisms by which normal cells can become cancerous. We identified 22 proteins with a regulatory role in EMT in OS whose endogenous functions impact these processes [53,54,62,64,78,91,97,100,112,116,126,130,131,139,140,141,142,143,144,145,146,151]. This group can be represented by several ubiquitin ligases [142,143] and deubiquitinases [97,145,146] that are known to target proteins critical for cell growth, proliferation, and differentiation. These were all found to be upregulated in clinical samples of osteosarcoma, and the overexpression or inhibition of these proteins was found to correlate with markers for EMT and OS cell proliferation.
The importance of immunity and inflammation on cancer progression is widely recognized [152], and these systems have also been implicated in the regulation of EMT [153]. Ren et al. found that PD-L2 knockdown decreased EMT and inhibited migration, invasion, and colony formation of OS cells in vitro, and reduced OS metastases in vivo in a mouse model [137].
Many of the described proteins are implicated in cell signaling [55,93,94,108,113,126,129,134,135,139,148]. In addition to cell–cell interactions, this broad grouping includes the regulation of multiple cell processes that affect multiple other pathways and functions, including but not limited to cell cycle regulation, inflammation, immunity, and metabolism.
Finally, a subset of the proteins associated with EMT in OS are either structural proteins or regulate cell structure via interaction with the cytoskeleton [89,106,121,122,129,148]. This is perhaps the simplest and most logical grouping given the key morphological changes that take place during the EMT transformation, as depicted in Figure 1. Interestingly, Yuan et al. found that Erythrocyte Membrane Protein Band 4.1-like 3 (EPB41L3)—a cytoskeletal protein involved in cytoskeletal rearrangement, intracellular transport, and signal transduction—was increased in OS tissues and cell lines but was associated with an inhibition of EMT, migration, invasion, and cell viability in OS cell culture [129]. This pattern of expression was opposite to all of the other proteins impacting EMT in OS identified in this review.
When reported, the EMT pathways most implicated in these studies were wnt and PI3K/Akt. The nuclear localization and, therefore, transcriptional activity of the wnt/β-catenin pathway has also been shown to be regulated by PI3K [30], suggesting overlap in these EMT control mechanisms. The most frequently identified downstream target was Snail, which is known to promote EMT by suppressing E-cadherin expression [155], and further upregulates wnt signaling and EMT [20].
6. Regulation of EMT in OS—Non-Coding Ribonucleic Acids
Another key group of regulatory factors of EMT/MET in OS are non-coding ribonucleic acids (ncRNAs). These molecules have many forms and functions [156], one of which is gene regulation. Typically identified through queries to the Gene Expression Omnibus (GEO), the differential expression of multiple separate long non-coding RNAs (lncRNAs) [157], microRNAs (miRNAs) [158], circular RNAs (circRNAs) [159,160], and pseudogenes [161] have been found to relate to OS prognosis [162]. Again, they were found to have a role in either promoting [57,63,66,76,85,86,90,95,98,99,101,103,109,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196] or inhibiting [47,49,51,65,77,92,96,102,104,111,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225] EMT and invasive cellular behaviors of OS cells in vitro, and there was significant overlap in the affected pathways and ultimate downstream targets. Very frequently there are multiple non-coding RNAs involved in the same pathway as they can also regulate other nucleic acids.
Unlike the pattern observed in the majority of these findings, Yuan et al. found that although erythrocyte membrane protein band 4.1-like 3 (EPB41L3) was upregulated in OS cell lines and clinical tissue samples, knockdown of EPB41L3 significantly increased the migration and invasion capacity of the investigated cell lines despite decreased cell viability [130]. The findings were similarly mixed for lncRNA NKILA [201] and miR-let-7d [225]. These studies highlight the complexity of EMT regulation in OS and suggest that it is only one possible factor relating to tumor behavior and prognosis.
7. Regulation of EMT in OS—The Tumor Microenvironment
There has been increased recognition of the importance of the tumor microenvironment on various cellular functions and characteristics. This is the three-dimensional structure surrounding tumor cells and comprises immune cells, vascular network, and extra-cellular matrix (ECM), among other components. The tumor microenvironment is unique not only for different cancer types but also for individual patients, and it is influenced by multiple factors, including patient sex and presence of metastases [226]. A better understanding of the interactions within the tumor micro-environment is expected to lead to the development of personalized treatments targeted at individual patients’ tumors.
Han et al. found that the presence of tumor-associated macrophages (TAMS) and the expression of the inflammatory marker cyclo-oxygenase 2 (COX2) correlated with OS metastases in clinical samples [80]. They also found co-culture of OS cells with TAMS promoted EMT and aggressive cellular features in vitro, which was reversible by COX2 inhibition. Additionally, COX2 inhibition reduced pulmonary metastases in vivo in a murine model [80]. Ling et al. found that Von Willebrand Factor (VWF)—which is secreted by the endothelial cells lining blood vessels—promoted EMT in vitro following OS and endothelial cell co-culture, as well as tumor growth and metastasis in vivo in a mouse model [227].
In addition to the cellular and biochemical makeup of the tumor microenvironment, the biomechanical properties of the ECM may also play a role in regulating EMT. Dai et al. developed a three-dimensional cell culture model with varying degrees of ECM stiffness [228]. This may be of particular relevance when evaluating OS tumors that exist in the bone—a relatively rigid environment—but eventually expand into the surrounding soft tissues, which are substantially less rigid. It may also account for some of the differences in OS metastatic patterns as more than 85% of metastatic OS occurs in the lungs, a soft tissue, compared to only 21% that occurs in the bone [229].
This entry is adapted from the peer-reviewed paper 10.3390/biom13020398
This entry is offline, you can click here to edit this entry!