Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Peripheral nerve injuries requiring surgical repair affect over 100,000 individuals in the US annually. Three accepted methods of peripheral repair include end-to-end, end-to-side, and side-to-side neurorrhaphy, each with its own set of indications.
- peripheral nerve injury
- surgical repair
- nerve coaptation
- molecular mechanisms
- end-to-end coaptation
1. Introduction
Peripheral nerve injuries (PNIs) affect approximately twenty million people in the US with a combined 100,000 patients requiring peripheral nerve surgery in the US annually [1,2]. Peripheral nerve injuries are commonly caused by motor vehicle accidents and penetrating injuries, frequently involving working-age adults under 40, imparting a significant work force burden [3,4]. The current surgical gold standard is a direct end-to-end (ETE) repair; however, this approach is only viable if the resulting gap is small and amenable to a tension-free repair [1,2,5]. Historically, the most common direct repair technique is end-to-end coaptation, indicated when nerve segments can be approximated without tension in the presence of a viable proximal nerve stump [6,7,8,9]. In cases where direct repair is not feasible, autografts and/or cadaveric allografts are often employed to fill the gap. Coaptation of the nerve graft with the proximal and distal nerve stump is similarly done in and ETE fashion. In the absence of a proximal nerve stump, such as in nerve root avulsions, other techniques such as end-to-side repair or less frequently side-to-side repair is utilized to avoid the need for donor nerve sacrifice [6].
2. Molecular Events Preceding Nerve Regeneration
Prior to discussing nerve coaptation techniques and the molecular mechanisms behind each of these, it is important to consider the general events occurring after nerve damage. Peripheral nerves consist of bundles of axons sheathed in three layers of connective tissue: epineurium, perineurium, and endoneurium (Figure 1). Each axon is enveloped by the endoneurium and Schwann Cells (SCs), the main glial cells of the peripheral nervous system. Groups of axons are organized into a fascicle by perineurium, and these fascicles are finally sheathed in epineurium to form the peripheral nerve. According to the Sunderland classification, peripheral nerve injuries are classified into five degrees depending on the mechanism and intensity of injury. These include segmental demyelination (first degree); axonal damage with intact endoneurium (second degree); axonal damage with endoneurium damage (third degree); axonal, endoneurial and perineurial damage (fourth degree); and loss of continuity of entire nerve trunk (fifth degree) [1,2]. For the purposes of this paper we will be discussing fifth degree nerve injury and regeneration. Damage to the axonal membrane results in two distinct temporal phases, a rapid phase consisting of rapid influx of sodium and calcium, and a slow phase characterized by activation and the retrograde transport of signaling molecules. The rapid influx of sodium and calcium activates calpain, leading to the sealing of the injured nerve membranes. It also generates a bust of action potentials at the proximal stump that is propagated to the soma via L-type voltage-gated calcium channels and the release of calcium from the endoplasmic reticulum. This results in the activation of the second messenger cAMP, which activates several important pro-regenerative transcription factors including dual leucine zipper-bearing kinase (DKL; also known as MAP3K12) and CREB1 (cAMP-responsive element-binding protein 1) [7,8,9,10]. The slower phase of injury is mediated via protein kinases, including calcium/calmodulin-dependent kinase 2(CMAK2); mitogen-activated protein kinases (MAPK) such as Erk1 and Erk2; phosphatidylinositol 3-kinase (PI3K); protein kinase A (PKA); protein kinase C (PKC); and c-jun N-terminal kinase (JNK). Retrograde trafficking systems transport these to the cell soma which then activates transcription factors such as CREB (cAMP responsive element binding protein), c-Jun, and ATF-3 (activating transcription factor 3). Combination of these processes enable activation of regeneration associated genes (RAGs) converting the nerve from a neuro-transmitter state to a regenerative state. For instance, downstream of the injury-induced PI3K-GSK3 (glycogen synthase kinase-3 signaling pathway), upregulates and phosphorylates SMAD family member 1 (SMAD1), causing its accumulation in the nucleus where it interacts with histone acetyltransferase p300 (p300HAT) which causes the over-expression of genes encoding cytoskeleton proteins such as tubulin and actin. Additionally, transcription factors (Sox11, c-JUN); cell adhesion and guidance receptors; growth-associated protein-43 (GAP43); and neurotropic factors (NTFs) such as NGF (Nerve Growth Factor) and BDNF (Brain-Derived Neurotropic Factor) and their receptors Trk, are also upregulated. There is a simultaneous downregulation of neurofilaments, neuropeptides and neurotransmitters gene expression, thus inhibiting the neurotransmitter function of the neuron. It also results in the ‘cell body response’ that is seen after PNI and includes swelling of the neuronal cell; chromatolysis (scattering of Nissl bodies or rough endoplasmic reticulum); migration of the nucleus to the peripheral of the neuron; and increased protein synthesis for regeneration [7,8,9,10,11,12,13,14,15].
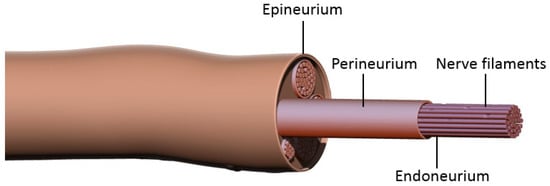
Figure 1. Peripheral nerve structure. Each axon is enveloped by endoneurium and Schwann cells. Groups of these nerve filaments are organized into fascicles by perineurium, and these fascicles are finally sheathed in epineurium to form a peripheral nerve.
Distally, the axons and myelin in the distal stump degenerate and are cleared (via a cascade of molecular and cellular events known as Wallerian degeneration (WD) (Figure 2) [16,17]. WD is a crucial process as the debris generates inhibitory signals such as myelin-associated glycoprotein that prevent axonal nascent neural growth from the proximal stump. SCs play a key role in the distal segment during WD. Axonal degeneration causes the loss of axonal contact with their SCs causing these adaptable cells (i.e., SCs) to re-enter the cell cycle and de-differentiate from a supportive to a repair phenotype. Gene expression of structural proteins such as myelin basic protein and myelin-associated glycoprotein are reduced, while reparative proteins such as NGF, basic fibroblast growth factor, Neurotrophin-3 (NT-3), neural cell adhesion molecule (NCAM), and glial maturation factor-β gene expression is upregulated. The de-differentiated SCs phagocytize degenerated myelin and repurpose it for future remyelination. They also express chemokine C-C motif ligand 2 (CCL2), leukemia inhibitory factor (LIF), IL-1α, IL-1β, IL-6, and pancreatitis-associated protein III (PAP-III) through the leaky nerve-blood barrier to recruit immune cells for further debris clearance [13,14,15,16,17]. Among immune cells, the first responders to the injured area are neutrophils, followed by macrophages that arrive 2–3 days after injury and peak at one week [16]. Macrophages clean up myelin via phospholipase A2 (PLA2) initiated phagocytosis, whose products, lysophosphatidylcholine (LPC) and C-reactive protein, activate the classic complement pathway [16]. In addition to cleaning up the debris, macrophages contribute to neural regeneration by producing proteases and growth-promoting factors, stimulating ECM remodeling, and regulating SCs. In addition to cleaning up the debris, macrophages contribute to neural regeneration by producing proteases and growth-promoting factors, stimulating ECM remodeling, and regulating Schwann cells [16].
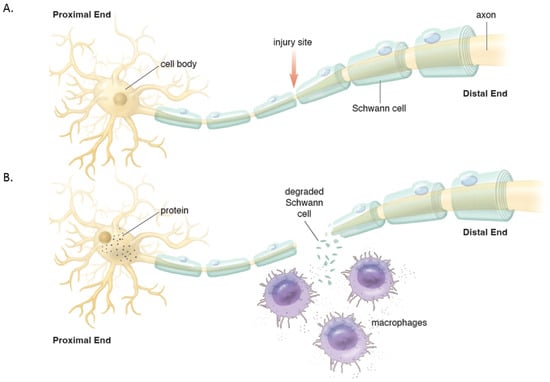
Figure 2. (A). Intact peripheral nerve. (B). Nerve transection leads to events in the proximal stump and distal stump. Proximally, the cell nucleus moves to the periphery and Nissl bodies disperse with increased protein synthesis to help repair the damage and seal the proximal membrane. Distally, Wallerian degeneration occurs which includes de-differentiation of Schwann cells, recruitment of macrophages that help breakdown and clear the debris in preparation of axonal regeneration.
3. End-to-End Repair
Early peripheral regeneration studies by Cruikshank in the late 18th century were instrumental in the ultimate surgical repair of nerves. Von Langebeck performed one of the earliest successful surgical nerve repairs of the median nerve in 1876, with return of function in a year [2,18,19]. Woolsey was first to describe the end-to-end suture technique in 1907 which led to the end-to-end (ETE) surgical repair being more acceptable in clinical practice [18,19]. World War I accelerated the developments in the field of nerve repair and yielded procedures such as nerve grafting, described by Tinel and Elsberg [16]. As nerve grafting became more popular, Tinel, Elsberg, and Babcock detailed surgical decision making and when end-to-end repair is preferred to grafts [16]. In 1964, Smith popularized the use of the microscope for peripheral nerve surgery, which enabled more advanced nerve repairs and led to the modern surgical nerve repair techniques [18,19].
Primary end-to-end neurorrhaphy (ETE), is the current standard for nerve repair if the repair can be performed in a tension-free manner [18]. If a tension-free primary repair is not possible a nerve graft (autograft vs. allograft) is utilized to bridge the gap and coapted to the proximal and distal nerve stumps using the ETE technique. Several end-to-end repair techniques have been attempted, including epineural, fascicular, and group-fascicular repair (Figure 3) [2]. Epineurial repair involves placing microsutures through the epineurium without traumatizing the fascicles. It has the advantage of reducing suture material between the two ends and therefore does not hinder axonal regeneration. It is also technically easier to perform and does not damage axons. Fascicular repair attempts to minimize the misdirection of fibers by allowing for direct coaptation of fascicles, but is technically extremely challenging, requires a greater number of microsutures through the perineurium, and has a higher chance of axonal damage. The additional sutures also tend to cause more foreign body reaction and scarring, hindering axonal regeneration. Group-fascicular repair entails repair of groups of fascicles rather than single fascicles and reduces fascicular injury and scarring, however it still creates additional tissue damage and foreign body reaction with additional sutures when compared to epineural repair [2,3]. Thus, the epineural repair (Figure 3A) has emerged the current gold standard for ETE nerve coaptation since it has the least interfascicular foreign body reaction, least scaring and best functional outcomes of these three techniques [2,18,19,20].
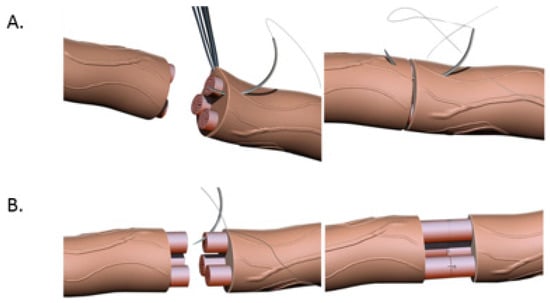
Figure 3. (A). Nerve coaptation via epineural suturing technique demonstrating sutures in the epineurium. (B). Nerve coaptation via fascicular repair demonstrating sutures in individual fascicles.
Timing and distance of the nerve injury from the target muscle are extremely important considerations while repairing peripheral nerve injuries. In general, the optimal timing for performing nerve repair is generally accepted to be within 72 h of injury, with acceptable outcomes up to 7 days after injury [2,21]. There are three time-dependent events that occur after nerve injury: (1) Wallerian degeneration; (2) the rate of nerve regeneration; and (3) motor end plate (MEP) degeneration that occurs after prolonged denervation. Axonal regeneration only commences once Wallerian degeneration is completed which takes 3–4 weeks to be completed. Once WD is complete, axonal regeneration begins at the rate of 1 mm/day or 1 inch/month [13]. Additionally, MEPs irreversibly degenerate at 12–18 months after injury if not reinnervated by then, due to the loss of nerve stimulation. Hence with proximal nerve injuries that are over 18 inches proximal to the target muscle (for instance neck injuries or brachial plexus injuries), even if the nerve coaptation is performed immediately after injury, it is destined to fail since the regenerating axons will only reach the MEPs after 18 months, by which time the MEPs have already irreversibly degenerated. Similarly, if the repair is performed after 12–18 months, there would not be any effective motor functional recovery, making it imperative to repair nerve injuries as soon as possible, especially in proximal injuries [2,13,20,21]. Hence for proximal injuries or delayed presentations, nerve transfers are utilized wherein a donor nerve (from a less useful synergistic muscle) closer to the denervated (but more useful) muscle is coapted primarily (i.e., ETE) to rescue denervated MEPs. This does require the sacrifice of a healthy donor motor nerve, which denervates a healthy, albeit less useful muscle. Sensory nerves are far more forgiving with regards to return of function after delayed or proximal repairs. ETE nerve coaptations are the most utilized surgical technique for PNIs clinically, however, there are several important issues to consider while employing this technique. ETE works best with a tension-free anastomosis, i.e., the proximal and distal end can be brought together without any tension on the nerve. Additionally, it needs a proximal nerve stump to be available, which as discussed above, is not possible for nerve avulsion injuries where the nerves are avulsed from their nerve roots in the spinal cord.
In end-to-end repairs, the injured axon in the proximal nerve segment elongates giving rise to axonal sprouts with a highly mobile tip known as the growth cone. The growth cone can assume different shapes to survey their environment via membrane protrusions called filopodia and lamellipodia [20]. Growth cones guide the axons into the distal nerve segment along the gradient of neurotropic cues in the microenvironment, including extracellular matrix, guidance receptors, cell adhesion molecules and NTFs [21,22]. Axonal growth thus occurs in three stages: protrusion, driven by filamentous actin (F-actin); engorgement, led by microtubule transport of organelles into the region; and consolidation, which occurs when the proximal growth cone is stabilized [23,24]. This process is tightly governed by molecular mechanisms that we explore in this section and are summarized in Figure 4. As previously mentioned, neuronal injury triggers a series of signals that ultimately alter gene expression within the injured neurons [11]. One mechanistic component involves the downregulation of myelin-differentiation genes: myelin transcription factor Egr2 (Krox20), cholesterol synthesis enzymes, P0 structural protein, myelin basic protein (MBP), and membrane associated proteins, such as myelin associated glycoprotein (MAG) and periaxin [25,26]. These steps are important to remove inhibitory signals, allowing the axonal sprouts to reach their distal sites. Subsequently, genes that are involved in the recruitment of pre-myelinating Schwann cells are up-regulated, notably L1, neural cell adhesion molecules (CAMs), p75 neurotrophin receptor (p75NTR), and glial fibrillary acidic protein (GFAP) [25,26,27]. Upregulation of RAGs (also known as growth-associated genes) in SCs is transient but is another essential step that includes secretion of NTFs, such as BDNF, glial derived neurotrophic factor (GDNF), nerve growth factor (NGF), artemin (Artn), neurotrophin-3 (NT3), VEGF, and ciliary neurotrophic factors (CNTF) (Figure 4) [11,26,27,28,29,30]. Of note, CNTF promotes phosphorylation of transcription factor STAT3 which is retrogradely transported to the neuron soma and is essential for axonal regeneration and neuronal protection against apoptosis. In addition to neurotrophic factors, cytoskeletal proteins undergo qualitative and quantitative changes characterized by an increase in actin, peripherin and tubulin that are essential for growth cone adhesion, and a decrease in neurofilaments. Basal lamina proteins, such as laminin allow SCs to interact with the growth cone adaptor molecules to promote axonal growth into the endoneurial tubes of the distal nerve stump following end-to-end repair [7,12,31,32]. Clinically, the IKVAV motif of laminin has been used in stem cell therapy for peripheral nerve injury due to its ability to guide SC migration [33,34,35]. Changes in SC gene expression are controlled by factors like c-Jun, which induces a cascade involving a transient upregulation of cyclin-dependent kinase Cdc2, in turn phosphorylating vimentin and allowing it to interact with basal lamina proteins [12,36,37,38,39]. Guidance molecules such a Netrin-1, Ephrin B2 and Slit3 are also upregulated at the injury site within the fibrous bridge that forms across the coaptation site after ETE coaptation. Macrophages secrete high levels of Slit3, while fibroblasts within the bridge and the advancing axons secrete Robo1 receptors their surface. The Slit3-Robo1 signaling axis acts as a restrictive signal to keep the axons within the nerve bridge. Netrin-1 expression on SCs has a dual action on endothelial cells (ECs) and the growth cone. It interacts with CD146 receptors on ECs, boosting their proliferation and migration, while it binds with DCC receptor (Deleted in Colorectal Cancer) on advancing axons, guiding the growth cone across the injury site [35].
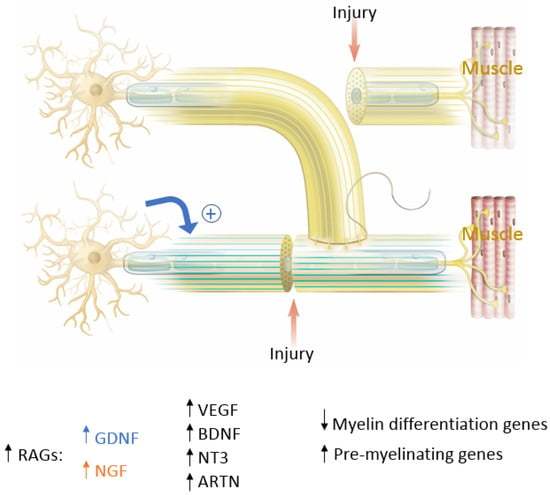
Figure 4. The key molecular players in peripheral nerve regeneration. In blue are factors that favor End-To-End (ETE) repair and in orange are factors that favor End-To-Side (ETS) repair. Factors that favor both ETE and ETS repair are in black. RAGs= Regeneration Associated Genes; GDNF = Glial Derived Neurotrophic Factor; NGF = Nerve Growth Factor; VEGF = Vascular Endothelial Growth Factor; BDNF = Brain-Derived Neurotropic Factor; NT3 = Neurotrophin-3; ARTN = Artemin.
Thus, extracellular matrix proteins, NTFs, and RAGs expressed by SCs are crucial for successful axonal growth, and if any component is missing, regeneration will not occur [22]. Notably, these changes in cellular expression are transient, and declines further if neurons are not in contact with their target, the distal segment [16]. This has significant clinical implications. Firstly, it explains why surgical nerve coaptations need to be performed within 72 h if possible. Secondly, it is essential to freshen the nerve ends prior to coaptation so as to prime the neurons to express the optimal RAGs. Freshening the nerve endings also helps tip the balance between the M1 and M2 macrophage polarization towards nerve regeneration. A shift towards M1 state and a lack of M2 macrophages leads to chronic inflammation and impaired nerve regeneration. M2 macrophages are extremely important to nerve regeneration due to their ability to response to injury-induced hypoxia. Hypoxia triggers hypoxia-inducible factor 1 alpha (HIF-1α) production in macrophages which is followed by an increased expression of VEGF-A (Vascular Endothelial growth factor A). This in turn increases EC migration and angiogenesis across the coaptation site, that guide SCs across to form bands of Büngner across the injury site. These bands are crucial for axonal guidance as axons march across the gap from the proximal to the distal nerve segment [16].
4. Muscle Reinnervation
In normally innervated muscles, motor branches (motoneurons) do not branch until they reach their destined muscle. Once intramuscular, they branch out with each motoneuron innervating a discrete territory in the muscle cross-section in the form of a ‘mosaic’ pattern [62]. Each of these functional units are known as a motor unit. With partial nerve injuries, the mosaic pattern of distribution is replaced by a “clumping” pattern, in an inverse ratio, i.e., lower the number of intact Mus, higher the clumping pattern. After complete nerve injury and end-to-end repair and reinnervation, the normal mosaic pattern is completely replaced by a “clumped” distribution. This occurs because of two reasons: firstly lower number of motoneurons are reaching the muscle and therefore compensate by capturing a higher number of muscle fibers in the vicinity with a resultant increased size of each MU, and a clumped pattern of muscle capture. Secondly since perisynaptic SCs present at the endplate region of the neuromuscular junction help guide regenerating axons to the end plate after nerve repair, regenerating axons miss “branch points”, being guided to the motor end plates by neutrophic factors generated by these presynaptic SCs. Thus, branching occurs only after they reach the muscle, accounting for the “clumping’ pattern. In general there is a 5–8 fold limit to this compensation mechanism, i.e., each motoneurons can only capture 5–8 fold more denervated muscle fibers. Thus, when less than 20% motoneurons reinnervate the denervated muscle, the entire muscle cannot be recaptured and muscle force declines [63]. Lack of muscle reinnervation can also occur secondary to misdirection since most developmental axon guidance cues are lost in adults. Axons can randomly enter vacant endoneurial tubes distally with sensory to motor and vice versa cross innervation. Alternatively motoneurons reinnervate different muscles than their native muscle [7].
Henneman’s size principle describes the relationship between the size of the motoneurons with the muscle fibers they innervate i.e., motoneurons with large cell bodies innervate fast-twitch, less fatigue-resistant muscle fibers, whereas motoneurons with small cell bodies innervate slow-twitch, higher fatigue-resistant fibers. These size relationships are lost after nerve transection and repair but return after axonal regeneration and muscle reinnervation. The size of nerves proximal to the site of transection and repair decreases after axotomy. Denervated muscle fibers also atrophy prior to their reinnervation. As reinnervation occurs, muscle fibers increase in size and nerve fibers recover their normal size. However, since the regenerating axons capture new muscle territory, it is the size-dependent branching of the regenerating nerves in the denervated intramuscular nerve sheaths and therefore the increase in MU size rather than then the cell body size that restores the MU forces and Henneman’s size principle [7].
As mentioned in above sections, after nerve injury, motoneurons undergo early and late changes in gene expression switching them to a regenerative pheonotype [63]. One intriguing aspect of this response is the intense shedding of synapses especially those of glutamatergic origin from motor cell bodies in the ventral horn of the spinal cord. Alvarez et al. distinguished this neuronal plasticity of motoneurons into two types: (1) a faster transient loss of synapses over the cell bodies of axotomized motoneurons that affects all types of synapses. And (2) a slower but permanent change in spinal cord circuitry that not only permanently affects axotomized motoneurons but other targets in the ventral horn, affecting both cell bodies and dendritic arbors, thus reconfiguring ventral horn motor circuitries to function after regeneration without direct proprioceptive or sensory feedback from muscle [63]. This process is modulated by injury severity suggesting a correlation between poor regeneration with sensory and/or motor targeting errors in the periphery that would render the previous central circuitries non-functional. Peripheral targeting errors thus must necessarily scramble motor circuit organization in the spinal cord rendering them dysfunctional. Thus, after nerve regeneration, the ventral horn operates without feedback about muscle length or motor output causing permanent changes in motor function. The extent to which the loss of these synaptic inputs further worsens peripheral function versus optimizes central circuits to the vagaries of jumbled peripheral targeting errors is unknown and currently under investigation.
This entry is adapted from the peer-reviewed paper 10.3390/jcm12041555
This entry is offline, you can click here to edit this entry!