Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Alzheimer’s disease (AD) is considered the most prevalent neurodegenerative disease and the leading cause of dementia worldwide. Sphingolipids, such as ceramide or sphingosine 1-phosphate, are bioactive molecules implicated in structural and signaling functions. Metabolic dysfunction in the highly conserved pathways to produce sphingolipids may lead to or be a consequence of an underlying disease.
- Alzheimer’s disease
- β-amyloid
- ceramide
- metabolism
1. Introduction: Alzheimer’s Disease and Sphingolipids
Alzheimer’s disease (AD) is considered the most prevalent neurodegenerative disease and the leading cause of dementia worldwide [1]. Based on clinical symptoms, the disease is characterized by memory loss (especially of recently learned information), temporal disorientation, language and personality disturbances, aggressiveness, agitation, and hallucinations, among others [2]. According to the World Health Organization (WHO), 55 million people suffer from dementia worldwide, with approximately 10 million new cases diagnosed each year [1,3]. In 2019, approximately 1.62 million people died from dementia, making it the seventh cause of death, and the fourth one in people over 70 years of age [4]. In addition, it is estimated that AD involves an annual cost of USD 1 trillion worldwide, which, together with the exponential increase in the number of cases due to the aging of the population, represents a great burden on healthcare systems [1].
The major neuropathological hallmarks of AD are the extracellular deposits of β-amyloid (Aβ) forming amyloid plaques as well as the intracellular accumulation of hyperphosphorylated tau protein in neurofibrillary tangles (NFT) [5]. In addition, AD is also characterized by vascular alterations [6], neuroinflammation [7], oxidative stress [8], and alterations in lipid and sphingolipid (SL) metabolism [9], among others.
The involvement of lipid metabolism in AD progression is well established. In this regard, the main genetic risk factor for sporadic late onset-AD is the presence of the ε4 allele of the apolipoprotein E gene (APOE4) [10]. There are different isoforms of APOE; APOE3 (which does not affect AD) is the most abundant, APOE2 (which appears to be beneficial for AD) is the least abundant; and APOE4 which is linked to the onset of AD by increasing the risk of developing the disease. Interestingly, ApoE is highly expressed in the brain. ApoE plays a critical role in cholesterol and other lipids’ transport and redistribution [11,12]. In addition, ApoE is also involved in other cellular processes such as cytoskeleton assembly and stability, mitochondrial function and integrity, and dendrite function and morphology [13]. Specifically, ApoE4 associates with Aβ more rapidly and stably than the other ApoE isoforms, facilitating the formation of amyloid plaques. Interestingly, ApoE4 mediates other pathological functions related to AD; for example, it stimulates tau phosphorylation, inhibits neurite outgrowth, and impairs neuronal plasticity and the integrity of the blood-brain barrier (BBB), among others [13]. Moreover, there are different AD-related single nucleotide polymorphisms (SNPs) related to plasma lipid levels. For example, FERM Domain Containing Kindlin 2 (FERMT2) and Membrane Spanning 4-Domains A6A (MS4A6A) show a significant differential association between AD patients and controls in all lipid classes (the association of MS4A6A with phosphatidylinositol (PI) levels is noteworthy); ATP Binding Cassette Subfamily A Member 7 (ABCA7) is associated with diglycerides (DG) and PI levels; Clusterin (CLU) with sphingomyelin (SM) and phosphatidylethanolamines (PE) levels; PI binding clathrin assembly protein (PICALM), Solute carrier family 24 member 4 (SLC24A4) and Sortilin-related receptor 1 (SORL1) with cholesteryl esters (ChE) levels; Bridging integrator 1 (BIN1) with triglycerides (TG) levels; and Complement C3b/C4b Receptor 1 (CR1) with PE levels [14]. Thus, the clear involvement of different types of lipids in AD is evident.
SLs are bioactive lipids with structural and second messenger functions. SLs participate in different cellular processes such as cell proliferation, migration, apoptosis, autophagy, senescence, and inflammation [15,16,17,18,19,20]. Moreover, SLs are highly expressed in the central nervous system (CNS), being the main component of the plasma membrane and myelin [21]. In addition, SLs have a strong relationship with different neurodegenerative diseases [9] and brain injury [22]. Among all, we highlight ceramide (Cer), the central metabolite in SL metabolism, and sphingosine 1-phosphate (S1P), the most studied. It is well known that Cer is a proapoptotic molecule, as it stimulates inflammation, autophagy, and oxidative stress by mitochondrial dysfunction [9,23], while its phosphorylated analog (ceramide 1-phosphate, C1P) or S1P stimulates cell survival, proliferation, and migration, among other functions [15]. S1P is also involved in neurodevelopment, synaptic transmission, neuroinflammation, and autophagy [9].
In this regard, genetic alterations in SL metabolism have been observed in brain tissue, cerebrospinal fluid (CSF), and the blood of patients with neurodegeneration [24,25,26,27]. Moreover, SLs have been associated with different pathological processes such as neuroinflammation and amyloid plaque formation, and thus are being proposed as promising biomarkers [28,29,30]. Consistently, mass spectrometry analysis of SL, called sphingolipidomics, has been performed to study the SL profile in brain samples as well as in biological fluids such as CSF, serum, or plasma. In fact, several studies have observed SL alterations in AD patients and patients with other neurodegenerative diseases compared to controls, and even in early clinical stages such as mild cognitive impairment (MCI) [31]. Moreover, these variations have been detected in different stages of AD progression, from prodromal to more advanced stages [31].
2. Sphingolipid Metabolism
Cer is considered the central metabolite of SL metabolism. So far, there are more than 30 described enzymes implicated in Cer metabolism. It is important to highlight that these enzymes are highly conserved and strictly regulated. The metabolism of SL can be differentiated into three main pathways, depending on the origin of the metabolites and their cellular location (Figure 1).
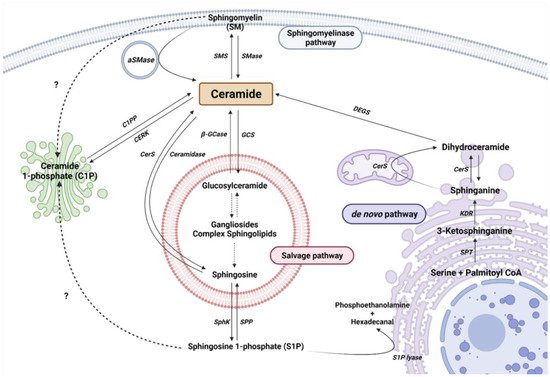
Figure 1. Sphingolipid metabolism. Single reactions are represented by solid arrows, while multiple-step reactions are shown as dashed arrows. Interrogation marks with dashed arrows point to possible mechanisms not yet described. Serine palmitoyltransferase (SPT), 3-keto-dihydrosphingosine reductase (KDR), ceramide Synthase (CerS) and dihydroceramide desaturase (DEGS), sphingomyelinase (SMase), acid sphingomyelinase (aSMase), sphingomyelin synthase (SMS), acid β-glucosidase (β-GCase), glucosylceramide synthase (GCS), ceramide synthase (CerS), sphingosine kinase (SphK), and sphingosine 1-phosphate phosphatase (SPP) are represented by their acronyms.
2.1. The De Novo Pathway
The de novo pathway takes place in the endoplasmic reticulum (ER). Serine palmitoyltransferase (SPT) is the first enzyme in the de novo pathway, which condensates palmitate and serine to produce 3-ketodehydrosphingosine (known also as 3-ketosphinganine). Being the first enzyme, it is of vital importance in controlling the rate of SL synthesis by de novo pathway. SPTLC1, SPTLC2, ssSPTa/b, and a negative regulatory subunit ORMDLs (homologs of the yeast and plant Orms) are the subunits that form the SPT complex in the ER membranes [32,33]. Subsequently, the 3-keto-dihydrosphingosine reductase (KDR) incorporates a hydrogen atom to 3-ketodehydrosphingosine, to synthesize sphinganine (Spha). Then, the ceramide synthase (CerS) enzyme introduces another acyl-CoA to form dihydroceramide (dhCer). So far, six isoforms of CerS (CerS1–6) have been described in mammals and plants [34]. Each isoform has a different affinity for Acyl-CoA molecules depending on the length of the chain. In this regard, CerS1 has more affinity to produce 18 carbon chain Cer (C18-Cer), CerS2 to C22/24-Cer, CerS3 to C26-Cer, CerS4 to C18/20-Cer, CerS5 to C14/16-Cer, and CerS6 to C14/16-Cer. Interestingly, C16-Cer synthesized by CerS6 is related to pro-survival events, while C18-Cer and longer are involved in pro-apoptotic pathways. Remarkably, CerS1 is highly expressed in the CNS. Finally, dihydroceramide desaturase (DEGS) introduces a double bond in position 4–5 trans of dhCer to form Cer.
2.2. The Sphingomyelinase (SMase Pathway)
SMase is a hydrolytic enzyme that degrades sphingomyelin (SM) at membrane level to generate Cer and phosphocholine in the cytosol, lysosomes, or lumen. So far, five isoforms have been described and classified according to their location, ionic regulation, and optimal pH activity. Acid SMase (aSMase) [35] is found in the lysosomes and plasma membrane [35]; its secreted form (Zn2+-dependent aSMase) is observed in saliva and serum [36]; neutral SMase (nSMase) has been detected in the nucleus, ER and plasma membrane [37]; and alkaline SMase (alkSMase) is secreted to intestinal tract lumen and human bile [38,39]. Oppositely, sphingomyelin synthase (SMS) catalyzes the production of SM from Cer. To date, three different SMSs have been described; SMS1, SMS2, and SMS-related protein (SMSr) [40]. It should be noted that different noxious stimuli can activate acid and neutral SMase and increase Cer synthesis, including different cytokines and interleukins, radiation, and cellular signals of cell cycle arrest or apoptosis [41].
2.3. The Salvage Pathway
The salvage pathway involves catabolic reactions to degrade complex SLs in lysosomes. Complex SLs are degraded through different reactions to produce Lactosyl-ceramide (LacCer). Then, LacCer hydrolase catalyzes the hydrolysis of LacCer to Glucosyl-Ceramide (GlcCer). Subsequently, GlcCer is hydrolyzed by acid β-glucosidase 1 (β-GCase) to form Cer. β-GCase is encoded by the GBA1 gene, whose deficiency or dysfunction produces GlcCer accumulation, leading to the development of the lysosomal storage disease known as Gaucher’s disease. In addition, mutations in GBA1 are associated with Parkinson’s Disease (PD) [42,43,44]. In the opposite direction, the glucosylceramide synthase (GCS) enzyme converts Cer into GlcCer. Cer can be transformed to sphingosine (Sph) by the ceramidase activity. Ceramidases are a small group of enzymes that differ in their optimal pH activity. Thus far, there are three alkaline enzymes, known as ACER1, ACER2, and ACER3, an acidic ceramidase (ASAH1), and a neutral ceramidase (ASAH2) [45,46]. Importantly, ASAH1 is ubiquitously found in lysosomal compartments. However, ASAH2 is observed in plasma membranes, being mainly expressed in the colon and small intestine [45]. Finally, Sph is secreted to the cytosol to produce Cer (by CerS in ER) or S1P.
2.4. Ceramide Kinase/Ceramide 1-Phosphate Phosphatase (CerK/CPP) and Sphingosine Kinase/Sphingosine 1-Phosphate Phosphatase (SphK/SPP) Axis
As explained, Sph can be phosphorylated to form S1P by sphingosine kinase (SK) activity. S1P is involved in inflammatory processes through its interaction with specific membrane receptors (S1PR), such as glia activation and vascular-mediated inflammation, among others [47,48,49]. Five S1P receptors (S1PR1–5) have been described [50]. Meanwhile, S1P phosphatase (SPP) or lipid phosphate phosphatase (LPP) catalyzes the dephosphorylation of S1P to Sph [19]. In addition, S1P can be catalyzed by the activity of S1P lyase to produce phosphoethanolamine and hexadecanal [51].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23158082
This entry is offline, you can click here to edit this entry!