Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Neurodegenerative disorders (NDDs) are major health issues in Western countries. Despite significant efforts, no effective therapeutics for NDDs exist. Several drugs that target epigenetic mechanisms (epidrugs) have been recently developed for the treatment of NDDs, and several of these are currently being tested in clinical trials.
- epigenetics
- neurodegeneration
- epinutraceuticals
1. Introduction
Neurophysiological mechanisms such as memory acquisition, learning, and motor coordination are, to a large extent, epigenetically regulated [32,33] (Figure 1). Alterations to the highly regulated epigenetic machinery increase the risk for the onset of various NDDs. These epigenetic aberrations target genes that are linked to synaptic plasticity, immune responses, cell development, and apoptosis [33,34,35,36,37].
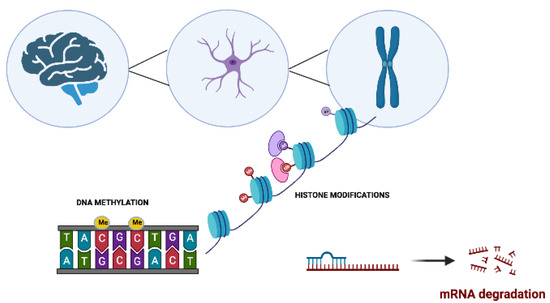
Figure 1. The main molecular epigenetic mechanisms in neurodegenerative disorders. These modifications include DNA methylation, histone modifications, and noncoding RNA-mediated alterations such as microRNA (miRNA) regulation.
2. Role of DNA Methylation in Neurodegenerative Disorders
DNA methylation levels are reduced in the AD and PD brain and in blood samples from animal models and human subjects with NDDs [38,39,40,41]. Downregulated DNMT and impaired vitamin B12 activities are the main factors that contribute to this global hypomethylation. Unlike patients with AD and PD, patients with amyotrophic lateral sclerosis (ALS) show increased DNMT expression and higher levels of DNA methylation than healthy individuals, suggesting that global DNA hypermethylation may be a contributing factor to the disease [42].
An increasing amount of data suggest a link between gene-specific methylation and neurodegeneration. A deficiency in vitamin B, for example, decreases glycogen synthase kinase 3β (GSK3β) methylation in AD patients; GSK3β expression is consequently increased, inducing tau phosphorylation, the formation of neurofibrillary tangles (NFTs), loss of cytoskeletal integrity, and cell death [43]. Genes such as bridging factor 1 (BIN1), complement receptor 1 (CR1), CD33, and tumor necrosis factor (TNFα), which are involved in cell death and neuroinflammation, are hypomethylated [44,45]. However, there are also several examples of DNA hypermethylation and decreased expression. Some of these genes include sortilin-related receptor (SORL1) and neprilysin (NEP), which are both involved in the degradation and clearance of Aβ. Other examples of hypermethylated genes are thromboxane A2 receptor (TBXA2R), sorbin, SH3 domain-containing 3 (SORBS3), and spectrin beta 4 (SPTBN4), which are hypermethylated in animal models of AD and in patients with AD [46]. Thus, blood DNA methylation has been proposed as a biomarker for dementia [38,39,41,47,48].
SNCA is a gene that encodes α-synuclein, a protein that is found in many tissues, including the brain. Mutations in the SNCA gene have been linked to several neurodegenerative diseases, most notably PD and Lewy body dementia. The promoter in SNCA is hypomethylated in blood and brain samples from PD patients, causing an overexpression of α−synuclein and fibrillary aggregation that promotes nigrostriatal degeneration. These patterns of hypomethylation and overexpression are associated with posttranslational modifications of α−synuclein and are also observed in the putamen and cerebral cortex of patients with sporadic PD [49].
3. Post-Translational Histone Modifications in Neurodegenerative Disorders
Histone modifications such as acetylation or methylation may contribute to the development and progression of NDDs. A correct balance between the activity of HAT (histone acetyltransferases) and that of HDAC (histone deacetylases) is essential for maintaining brain homeostasis [50]. Increased histone acetylation has been implicated in AD pathology, and recent data indicate that HDAC inhibitors are neuroprotective by regulating memory and synaptic dysfunctions in cellular and animal models of AD [51].
Alpha-synuclein binds to histones, prevents H3 acetylation, and induces neurotoxicity [52]. Tri-methylation of histone H3 on lysine 4 (H3K4me3) is increased in the SNCA promoter in the substantia nigra in patients with PD. H3K4me3 is a transcription-promoting histone modification that increases gene transcription and expression, suggesting that H3K4 tri-methylation is involved in the induction of α-synuclein overexpression [53]. Long-term treatment with levodopa also causes deacetylation of histone H4 at lysine 5, 8, 12, and 16. Exposure to the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) destroys dopaminergic neurons and causes PD-like symptoms, but it also increases the levels of histone H3 acetylation, which are reduced by treatment with levodopa [54]. High levels of histone H2A, H3, and H4 acetylation are present in dopaminergic neurons in post-mortem PD patients [55]. Indeed, treatment with HDAC inhibitors reduces α-synuclein neurotoxicity in neuroblastoma cells and in dopaminergic neurons in the α-synuclein transgenic Drosophila model of PD [52,56,57]. The SIRT family of proteins promotes lifespan and healthy aging by modulating a variety of cellular processes, including metabolism, chromatin silencing, cellular differentiation and stress response, inflammation, and cell death [57]. SIRT1, the best-studied member of the SIRT family, is an NAD-dependent protein deacetylase that regulates many important biological processes by removing acetyl groups from target proteins. In recent years, SIRTs have been implicated in neurodegenerative diseases such as AD and PD, and several studies show that SIRTs are neuroprotective [58].
Histone methyltransferases (HMTs) and demethylases (HMDs) catalyze the methylation and demethylation of histones, respectively, and have become an innovative target for treating or preventing NDDs. A dynamic balance between HAT and HMT regulates suppressive chromatin, which is associated with NDD pathology and progression [59]. In pre-plaque AD transgenic mice, H3K14 and H3K9me2 histone methylation levels are elevated [60], whereas during the progression of PD, the demethylase Jumonji domain-containing protein-3 (Jmjd3) is essential for modulating microglia phenotypes [61].
4. Regulation of micro-RNAs in Neurodegenerative Disorders
MicroRNAs (miRNAs) are a class of short noncoding RNA molecules that regulate the expression of genes involved in several cellular processes such as differentiation, proliferation, and cell death. They have been implicated in several neurodegenerative diseases, including AD, PD, and Huntington’s disease (HD), in which they are involved in neuroinflammation and cell death [62]. MiRNAs can be detected and quantified in peripheral biofluids, such as plasma, serum, and cerebrospinal fluid, and in peripheral blood mononuclear cells (PBMCs), suggesting that circulating miRNAs extracted from blood or other biofluids may serve as non-invasive and cost-effective biomarkers for the early detection of NDDs such as AD and PD. MiRNAs could therefore be utilized as a screening tool for the early detection and monitoring of NDD progression. In AD, miRNAs regulate synaptic activity, and several miRNAs (e.g., miR-124, miR-125b, miR-34c, and miR-132) are enriched in affected synapses [63,64]. Dysregulation of brain-specific miRNAs adversely modulates synaptic activity in AD by suppressing their target genes, thus impairing synaptic activation and transmission. This has pathogenic consequences such as neurotrophic and synaptic deficits and astrogliosis. Siedlecki-Wullich et al. (2019) showed that a plasma miRNA signature comprising miR-210-3p, miR-181c-5p, and miR-92a-3p may be effective as a non-invasive clinical biomarker for the diagnosis of AD, and which could be utilized to improve future treatment strategies [65]. MiRNAs may, therefore, be informative and specific biomarkers for evaluating the staging, progression, and prognosis of NDDs [45].
This entry is adapted from the peer-reviewed paper 10.3390/ph16020216
This entry is offline, you can click here to edit this entry!