Radiotherapy and, more recently, PARP inhibitors (PARPis) and immune-checkpoint inhibitors represent effective tools in cancer therapy. Radiotherapy exerts its effects not only by damaging DNA and inducing tumor cell death, but also stimulating anti-tumor immune responses. PARPis are known to exert their therapeutic effects by inhibiting DNA repair, and they may be used in combination with radiotherapy. Both radiotherapy and PARPis modulate inflammatory signals and stimulate type I IFN (IFN-I)-dependent immune activation. However, they can also support the development of an immunosuppressive tumor environment and upregulate PD-L1 expression on tumor cells. When provided as monotherapy, immune-checkpoint inhibitors (mainly antibodies to CTLA-4 and the PD-1/PD-L1 axis) result particularly effective only in immunogenic tumors. Combinations of immunotherapy with therapies that favor priming of the immune response to tumor-associated antigens are, therefore, suitable strategies. The widely explored association of radiotherapy and immunotherapy has confirmed this benefit for several cancers. Association with PARPis has also been investigated in clinical trials. Immunotherapy counteracts the immunosuppressive effects of radiotherapy and/or PARPis and synergies with their immunological effects, promoting and unleashing immune responses toward primary and metastatic lesions (abscopal effect).
- radiotherapy
- immunotherapy
- PARP inhibitors
- tumor immunity
- combined therapies
- cancer immunology
- immune checkpoints
1. Introduction
2. Radiotherapy
Radiotherapy plays a major role in the treatment of a wide range of malignancies. Around 60–70% of patients undergo treatments, mostly with photon therapy (X- or γ-rays), in addition to others with heavy ions and protons.
2.1. Immuno-Stimulating Effects of Radiotherapy
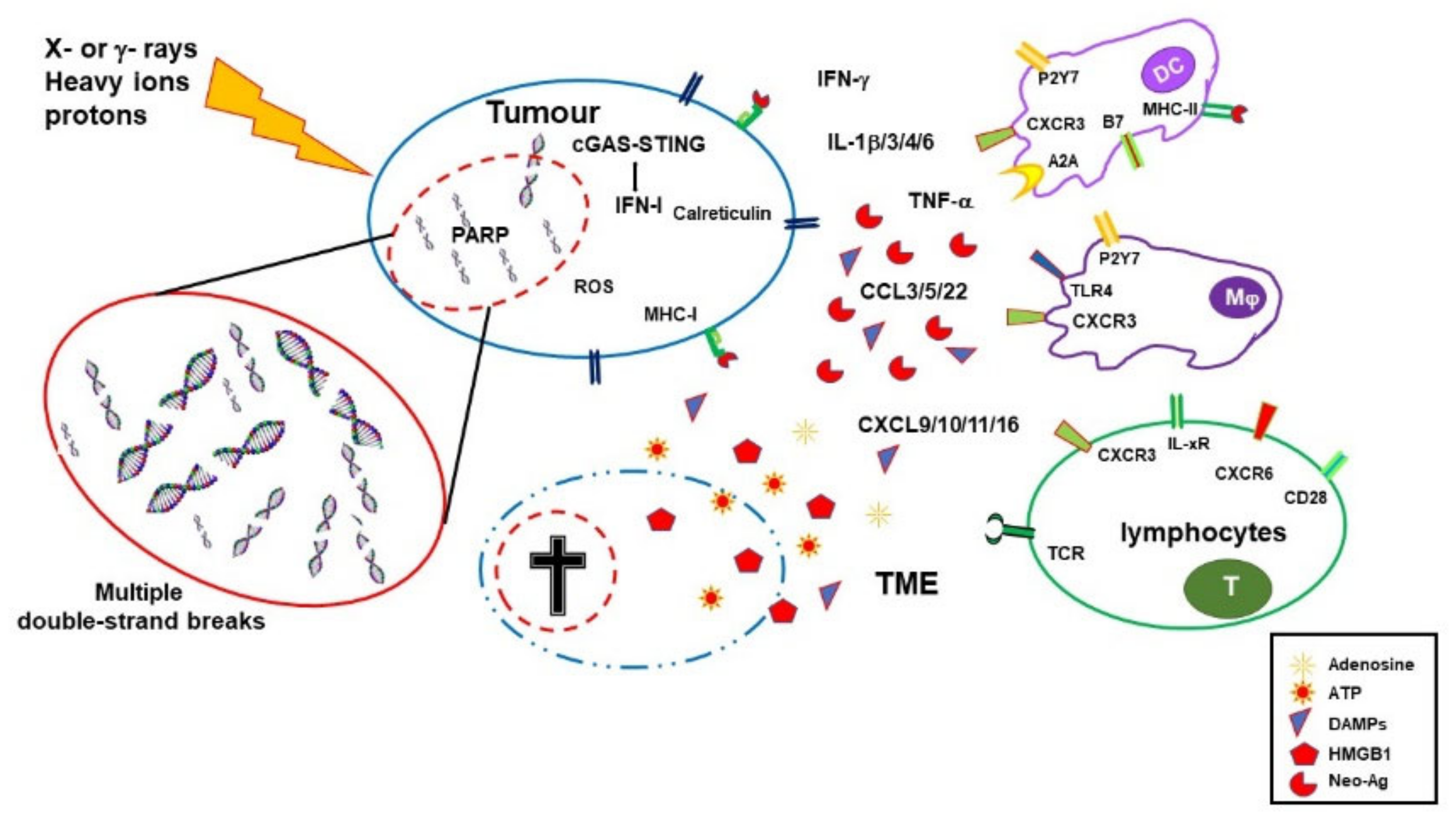
2.2. Immuno-Depressing Effects of Radiotherapy
3. PARP Inhibitors
PARP-1, the most abundant member of the poly(ADP-ribose) (PAR) polymerase (PARP) family, more recently defined as diphtheria toxin-like ADP-ribosyltransferases (ARTDs), accounts for the majority of PARylation activity and has a high DNA damage-sensing ability [40]. Free DNA ends activate PARP-1, which highly PARylates itself and detaches from chromatin. Indeed, addition of PARs radically changes the electric charge of the targeted molecule, rendering it highly negative. As a consequence, PARylated proteins are electrostatically repulsed by the DNA, a mechanism involved in chromatin accessibility to DNA repair enzymes (and to DNA transcription and replication regulators). PARP-1 also generates large amounts of PARs that work as scaffolds recruiting DNA repair enzymes to the lesion site, including XRCC1 [41]. PARP-1 plays a central role in orchestrating responses to genotoxic stress and represents a critical enzyme in single-strand break and alternative end-joining repair [42][43]. However, recent studies also indicated that PARP-1 plays a role in double-strand break (DSB) repair mechanisms, including homologous recombination and classical nonhomologous end-joining (c-NHEJ) [44][45].
Following a long period of preclinical and clinical studies, PARP inhibitors (PARPis) reached wide clinical use with the approval of olaparib (AZD-2281) in 2014 and later on of niraparib (MK-4827), rucaparib (AG-014699), talazoparib (BMN673), and veliparib (ABT888) for treatment of ovarian, breast, prostate, and pancreatic cancer [46][47]. PARPis are the first clinically approved drugs exploiting synthetic lethality; that is, they target a function specifically vital in mutation-bearing cancer cells [48][49]. PARPis were shown to be lethal in homologous recombination (HR)-deficient BRCA1/BRCA2-mutated cancers, likely because collapsed replication forks are no longer repaired [50][51]. However, recent preclinical and early clinical studies also sustained the use of PARPis in other molecular subsets of cancer, including cancers with high replication stress [52].
All clinically approved PARPis share a nicotinamide-based moiety that inhibits PARP-1 enzymatic activity by competing for binding to the catalytic site with NAD. PARPis prevent PARP-1 auto-PARylation and its consequent removal from chromatin and DNA lesions. This effect, termed PARP trapping, is currently the preferred interpretative model of the PARPis mechanism of action. Indeed, cytotoxicity due to PARP correlates with the ability to trap PARP on DNA lesions and is more cytotoxic than gene deletion. PARP trapping leads to replication fork collapse during the S phase and consequent cell death [53][54].
4. Synergy between PARPi and Radiotherapy
5. Synergic Immunological Effects of RT and PARPi
6. Immune-Checkpoint Inhibitors
7. Synergy between Radiotherapy and ICI
8. Synergy between PARPi and ICI
9. Perspectives: Triple RT, PARPi and ICI Combinations to Overcome Respective Limitations
As described above RT, PARPis and ICI have a certain therapeutic success when used alone but it is their combination that can result in a better and prolonged disease control. RT and PARPis synergize in inducing DNA damage and tumor cell death. They also induce immune stimulating factors potentially generating an immunogenic microenvironment and favoring immune infiltration. However, they also activate immune suppressive mechanisms and indeed the induction of a systemic immune response with abscopal effects remains uncommon and/or limited. On the other hand, ICIs can lower the threshold for immune activation, reinvigorate exhausted T cells, and dampen the action of regulatory T cells, consequently sustaining systemic immune responses and abscopal effect. Nevertheless, to be effective ICIs require a TME that allows priming of immune responses to tumor-associated antigens and tumor infiltration by leukocytes. Combinations of immunotherapy with therapies that favor priming of immune responses, such as RT, obtained important therapeutic success in clinical studies, with protocols including different forms of RT and ICI having been approved for several (advanced) cancers. Also the more recent association of PARPis and ICIs showed some clinical benefits. Altogether these results and the considerations expressed above encourage the use of combined therapies that include RT, PARPis and ICIs.
Promising results from initial studies in experimental models confirmed that the triple combination of RT, PARPis and ICI improve tumor infiltrate, and prime and unleash anti-tumor, T-cell-mediated, immune responses in mouse models [115][116]. Several phase I to III clinical trials, aimed at exploring different combinations of radiotherapy, PARPis and ICIs, included at least one arm with the concomitant or sequential use of these three therapeutic agents (often in addition to standard chemo-therapy). The effects of PARPis together with RT and ICI, targeting either CTLA-4 or PD-1/PD-L1 or both pathways, will be assessed in NSCLC, SCLC, breast, prostate, pancreatic, gastroesophageal, rectal, head and neck carcinomas. Many of these trials are still recruiting or not yet active. A wealthy of results will be available on these promising therapeutic combinations in forthcoming years (Table 1).
| Title | Conditions | Therapies | Phase | Estimated Enrollment (Patients) |
Status | Estimated Completion Dates | NCT Number | Last Update Posted |
|---|---|---|---|---|---|---|---|---|
| Testing the safety of the anticancer drugs durvalumab and olaparib during radiation therapy for locally advanced unresectable pancreatic cancer | Locally advanced pancreatic carcinoma Stage II or III pancreatic cancer Unresectable pancreatic carcinoma |
Durvalumab Olaparib RT |
I | 18 | Recruiting | Primary and final: 31 March 2024 | 05411094 | 1 December 2022 |
| A safety study adding niraparib and dostarlimab to radiation therapy for rectal cancers | Rectal neoplasms Rectal neoplasm malignant |
Niraparib Dostarlimab Short course RT |
I–II | 38 | Recruiting | Primary: 31 December 2024 Final: 31 December 2026 |
04926324 | 26 July 2022 |
| Niraparib + dostarlimab + RT in pancreatic cancer | Pancreatic cancer Metastatic pancreatic cancer |
Niraparib Dostarlimab RT |
II | 25 | Active, not recruiting |
Primary: 19 January 2022 Final: October 2026 |
04409002 | 8 September 2022 |
| Radiation, immunotherapy, and PARP inhibitor in triple-negative breast cancer | Breast cancer TNBC |
Niraparib Dostarlimab RT |
II | 32 | Recruiting | Primary: 1 April 2023 Final: 1 December 2029 |
04837209 | 23 December 2022 |
| Radiotherapy and durvalumab/durvalumab combo (tremelimumab/olaparid) for small-cell lung cancer | SCLC extensive stage SCLC |
Durvalumab Tremelimumab Olaparib Thoracic RT |
I | 25 | Active, not recruiting |
Primary and final: 1 June 2023 | 03923270 | 6 January 2023 |
| A study of radiation therapy with pembrolizumab and olaparib or other radiosensitizers in women who have triple-negative or hormone-receptor positive/HER2 negative breast cancer | TNBC Metastatic breast cancer |
Pembrolizumab Olaparib RT |
II | 34 | Recruiting | Primary and final: January 2025 | 04683679 | 21 October 2022 |
| Pembro with radiation with or without olaparib | Prostate cancer | Pembrolizumab Olaparib Androgen deprivation therapy RT |
II | 64 | Not yet recruiting |
Primary: 2 January 2025 Final: 2 January 2028 |
05568550 | 5 October 2022 |
| Olaparib and durvalumab with carboplatin, etoposide, and/or radiation therapy for the treatment of extensive-stage small-cell lung cancer, PRIO trial |
Extensive-stage SCLC Stage IV lung cancer Stage IVA lung cancer Stage IVB lung cancer |
Carboplatin Durvalumab Etoposide Olaparib RT |
I–II | 63 | Recruiting | Primary and final: 31 January 2024 | 04728230 | 9 November 2022 |
| Study of SBRT/olaparib followed by pembrolizumab/olaparib in gastric cancers | Gastric cancer Gastroesophageal cancer |
Pembrolizumab Olaparib SBRT |
II | 26 | Recruiting | Primary: December 2025 Final: December 2028 |
05379972 | 5 January 2023 |
| Placebo-controlled study of concurrent chemoradiation therapy with pembrolizumab followed by pembrolizumab and olaparib in newly diagnosed treatment-naïve limited-stage small-cell lung cancer (LS-SCLC) (MK 7339-013/KEYLYNK-013) | SCLC | Pembrolizumab (2 doses) Olaparib Etoposide Platinum Standard thoracic RT Prophylactic cranial irradiation |
III | 672 | Recruiting | Primary and final: 28 October 2027 | 04624204 | 23 December 2022 |
| Pembrolizumab plus olaparib in LA-HNSCC | Head and neck squamous cell carcinoma | Pembrolizumab Olaparib Cisplatin IMRT |
II | 45 | Recruiting | Primary: 31 October 2024 Final: 31 October 2025 |
05366166 | 28 October 2022 |
| Study of pembrolizumab with concurrent chemoradiation therapy, followed by pembrolizumab with or without olaparib in stage III non-small-cell lung cancer (NSCLC) (MK-7339-012/KEYLYNK-012) | Lung neoplasms NSCLC |
Pembrolizumab Olaparib Etoposide Carboplatin Cisplatin Paclitaxel Pemetrexed Thoracic RT Durvalumab |
III | 870 | Recruiting | Primary and final: 6 July 2026 | 04380636 | 30 November 2022 |
This entry is adapted from the peer-reviewed paper 10.3390/cancers15041093
References
- Sutherland, B.M.; Bennett, P.V.; Sutherland, J.C.; Laval, J. Clustered DNA damages induced by x rays in human cells. Radiat. Res. 2002, 157, 611–616.
- Goodhead, D.T. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int. J. Radiat. Biol. 1994, 65, 7–17.
- Lu, Z.; Zheng, X.; Ding, C.; Zou, Z.; Liang, Y.; Zhou, Y.; Li, X. Deciphering the Biological Effects of Radiotherapy in Cancer Cells. Biomolecules 2022, 12, 1167.
- Alterio, D.; Gugliandolo, S.G.; Augugliaro, M.; Marvaso, G.; Gandini, S.; Bellerba, F.; Russell-Edu, S.W.; De Simone, I.; Cinquini, M.; Starzyńska, A.; et al. IMRT versus 2D/3D conformal RT in oropharyngeal cancer: A review of the literature and meta-analysis. Oral Dis. 2021, 27, 1644–1653.
- Marta, G.N.; Silva, V.; de Andrade Carvalho, H.; de Arruda, F.F.; Hanna, S.A.; Gadia, R.; da Silva, J.L.; Correa, S.F.; Vita Abreu, C.E.; Riera, R. Intensity-modulated radiation therapy for head and neck cancer: Systematic review and meta-analysis. Radiother. Oncol. 2014, 110, 9–15.
- Byun, H.K.; Han, M.C.; Yang, K.; Kim, J.S.; Yoo, G.S.; Koom, W.S.; Kim, Y.B. Physical and Biological Characteristics of Particle Therapy for Oncologists. Cancer Res. Treat. 2021, 53, 611–620.
- Najafi, M.; Motevaseli, E.; Shirazi, A.; Geraily, G.; Rezaeyan, A.; Norouzi, F.; Rezapoor, S.; Abdollahi, H. Mechanisms of inflammatory responses to radiation and normal tissues toxicity: Clinical implications. Int. J. Radiat. Biol. 2018, 94, 335–356.
- Rodríguez-Ruiz, M.E.; Vanpouille-Box, C.; Melero, I.; Formenti, S.C.; Demaria, S. Immunological Mechanisms Responsible for Radiation-Induced Abscopal Effect. Trends Immunol. 2018, 39, 644–655.
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852.
- Yamazaki, T.; Galluzzi, L. Mitochondrial control of innate immune signaling by irradiated cancer cells. OncoImmunology 2020, 9, 1797292.
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.-X.; Auh, S.L. The Efficacy of Radiotherapy Relies upon Induction of Type I Interferon–Dependent Innate and Adaptive Immunity. Cancer Res. 2011, 71, 2488–2496.
- Du, J.; Kageyama, S.I.; Hirata, H.; Motegi, A.; Nakamura, M.; Hirano, Y.; Okumura, M.; Yamashita, R.; Tsuchihara, K.; Hojo, H.; et al. Comparative analysis of the immune responses in cancer cells irradiated with X-ray, proton and carbon-ion beams. Biochem. Biophys. Res. Commun. 2021, 585, 55–60.
- Cytlak, U.M.; Dyer, D.P.; Honeychurch, J.; Williams, K.J.; Travis, M.A.; Illidge, T.M. Immunomodulation by radiotherapy in tumour control and normal tissue toxicity. Nat. Rev. Immunol. 2022, 22, 124–138.
- Connolly, K.A.; Belt, B.A.; Figueroa, N.M.; Murthy, A.; Patel, A.; Kim, M.; Lord, E.M.; Linehan, D.C.; Gerber, S.A. Increasing the efficacy of radiotherapy by modulating the CCR2/CCR5 chemokine axes. Oncotarget 2016, 7, 86522–86535.
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425.
- Li, H.; Chen, X.; Zeng, W.; Zhou, W.; Zhou, Q.; Wang, Z.; Jiang, W.; Xie, B.; Sun, L.Q. Radiation-Enhanced Expression of CCL22 in Nasopharyngeal Carcinoma is Associated With CCR4(+) CD8 T Cell Recruitment. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 126–139.
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107.
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875.
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 20, 95–112.
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.M.; Michalak, M.; Murphy-Ullrich, J.E. Calreticulin: Non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010, 24, 665–683.
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334.
- Gameiro, S.R.; Malamas, A.S.; Bernstein, M.B.; Tsang, K.Y.; Vassantachart, A.; Sahoo, N.; Tailor, R.; Pidikiti, R.; Guha, C.P.; Hahn, S.M.; et al. Tumor Cells Surviving Exposure to Proton or Photon Radiation Share a Common Immunogenic Modulation Signature, Rendering Them More Sensitive to T Cell–Mediated Killing. Int. J. Radiat. Oncol. Biol. 2016, 95, 120–130.
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10.
- Procureur, A.; Simonaggio, A.; Bibault, J.E.; Oudard, S.; Vano, Y.A. Enhance the Immune Checkpoint Inhibitors Efficacy with Radiotherapy Induced Immunogenic Cell Death: A Comprehensive Review and Latest Developments. Cancers 2021, 13, 678.
- Mauch, P.; Constine, L.; Greenberger, J.; Knospe, W.; Sullivan, J.; Liesveld, J.L.; Deeg, H.J. Hematopoietic stem cell compartment: Acute and late effects of radiation therapy and chemotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1995, 31, 1319–1339.
- Frasca, D.; Guidi, F.; Arbitrio, M.; Pioli, C.; Poccia, F.; Cicconi, R.; Doria, G. Hematopoietic reconstitution after lethal irradiation and bone marrow transplantation: Effects of different hematopoietic cytokines on the recovery of thymus, spleen and blood cells. Bone Marrow Transplant. 2000, 25, 427–433.
- Frasca, D.; Pioli, C.; Guidi, F.; Pucci, S.; Arbitrio, M.; Leter, G.; Doria, G. IL-11 synergizes with IL-3 in promoting the recovery of the immune system after irradiation. Int. Immunol. 1996, 8, 1651–1657.
- Heylmann, D.; Rödel, F.; Kindler, T.; Kaina, B. Radiation sensitivity of human and murine peripheral blood lymphocytes, stem and progenitor cells. Biochim. Biophys. Acta 2014, 1846, 121–129.
- Berte, N.; Eich, M.; Heylmann, D.; Koks, C.; Van Gool, S.W.; Kaina, B. Impaired DNA repair in mouse monocytes compared to macrophages and precursors. DNA Repair 2021, 98, 103037.
- Leblond, M.M.; Pérès, E.A.; Helaine, C.; Gérault, A.N.; Moulin, D.; Anfray, C.; Divoux, D.; Petit, E.; Bernaudin, M.; Valable, S. M2 macrophages are more resistant than M1 macrophages following radiation therapy in the context of glioblastoma. Oncotarget 2017, 8, 72597–72612.
- Groves, A.M.; Johnston, C.J.; Misra, R.S.; Williams, J.P.; Finkelstein, J.N. Effects of IL-4 on pulmonary fibrosis and the accumulation and phenotype of macrophage subpopulations following thoracic irradiation. Int. J. Radiat. Biol. 2016, 92, 754–765.
- Arina, A.; Beckett, M.; Fernandez, C.; Zheng, W.; Pitroda, S.; Chmura, S.J.; Luke, J.J.; Forde, M.; Hou, Y.; Burnette, B.; et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat. Commun. 2019, 10, 3959.
- Qinfeng, S.; Depu, W.; Xiaofeng, Y.; Shah, W.; Hongwei, C.; Yili, W. In situ observation of the effects of local irradiation on cytotoxic and regulatory T lymphocytes in cervical cancer tissue. Radiat. Res. 2013, 179, 584–589.
- Qu, Y.; Jin, S.; Zhang, A.; Zhang, B.; Shi, X.; Wang, J.; Zhao, Y. Gamma-ray resistance of regulatory CD4+CD25+Foxp3+ T cells in mice. Radiat. Res. 2010, 173, 148–157.
- Zhai, D.; An, D.; Wan, C.; Yang, K. Radiotherapy: Brightness and darkness in the era of immunotherapy. Transl. Oncol. 2022, 19, 101366.
- Derer, A.; Spiljar, M.; Bäumler, M.; Hecht, M.; Fietkau, R.; Frey, B.; Gaipl, U.S. Chemoradiation Increases PD-L1 Expression in Certain Melanoma and Glioblastoma Cells. Front. Immunol. 2016, 7, 610.
- Gao, Y.; Li, Y.; Lin, Z.; Zeng, Y.; Huang, Z.; Han, L.; Zhong, Y.; Gong, Y.; Wu, Q.; Xie, C. Ataxia telangiectasia mutated kinase inhibition promotes irradiation-induced PD-L1 expression in tumour-associated macrophages through IFN-I/JAK signalling pathway. Immunology 2022, 168, 346–361.
- Mondini, M.; Loyher, P.-L.; Hamon, P.; Gerbé de Thoré, M.; Laviron, M.; Berthelot, K.; Clémenson, C.; Salomon, B.L.; Combadière, C.; Deutsch, E.; et al. CCR2-Dependent Recruitment of Tregs and Monocytes Following Radiotherapy Is Associated with TNFα-Mediated Resistance. Cancer Immunol. Res. 2019, 7, 376–387.
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017, 8, 1736.
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24.
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533.
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 711, 61–72.
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732.
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182.
- Ahmed, E.A.; Alzahrani, A.M.; Scherthan, H. Parp1-Dependent DNA Double-Strand Break Repair in Irradiated Late Pachytene Spermatocytes. DNA Cell Biol. 2021, 40, 209–218.
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533.
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763.
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917.
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921.
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771.
- Zandarashvili, L.; Langelier, M.F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368, eaax6367.
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443.
- Kupczyk, P.; Simiczyjew, A.; Marczuk, J.; Dratkiewicz, E.; Beberok, A.; Rok, J.; Pieniazek, M.; Biecek, P.; Nevozhay, D.; Slowikowski, B.; et al. PARP1 as a Marker of an Aggressive Clinical Phenotype in Cutaneous Melanoma—A Clinical and an In Vitro Study. Cells 2021, 10, 286.
- Raleigh, D.; Ahmed, K.M.; Zhang, H.; Ziaee, S.; Park, C.C. PARP-1 modulates β1-integrin/NF-κB-mediated radioresistance in human breast cancer. J. Cancer Ther. Res. 2016, 5, 1.
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly ADP-ribose polymerase inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179.
- Zhao, W.; Hu, H.; Mo, Q.; Guan, Y.; Li, Y.; Du, Y.; Li, L. Function and mechanism of combined PARP-1 and BRCA genes in regulating the radiosensitivity of breast cancer cells. Int. J. Clin. Exp. Pathol. 2019, 12, 3915–3920.
- Dungey, F.A.; Löser, D.A.; Chalmers, A.J. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-Ribose) polymerase: Mechanisms and therapeutic potential. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 1188–1197.
- Elser, M.; Borsig, L.; Hassa, P.O.; Erener, S.; Messner, S.; Valovka, T.; Keller, S.; Gassmann, M.; Hottiger, M.O. Poly(ADP-Ribose) Polymerase 1 Promotes Tumor Cell Survival by Coactivating Hypoxia-Inducible Factor-1–Dependent Gene Expression. Mol. Cancer Res. 2008, 6, 282–290.
- Gonzalez-Flores, A.; Aguilar-Quesada, R.; Siles, E.; Pozo, S.; Rodríguez-Lara, M.I.; López-Jiménez, L.; López-Rodríguez, M.; Peralta-Leal, A.; Villar, D.; Martín-Oliva, D.; et al. Interaction between PARP-1 and HIF-2α in the hypoxic response. Oncogene 2014, 33, 891–898.
- Rosado, M.M.; Bennici, E.; Novelli, F.; Pioli, C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 2013, 139, 428–437.
- Rosado, M.M.; Pioli, C. ADP-ribosylation in evasion, promotion and exacerbation of immune responses. Immunology 2021, 164, 15–30.
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Investig. 2019, 129, 1211–1228.
- Lim, J.Y.H.; Gerber, S.A.; Murphy, S.P.; Lord, E.M. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8+ T cells. Cancer Immunol. Immunother. 2014, 63, 259–271.
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737.
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1−/− murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556.
- Zhang, N.; Gao, Y.; Zeng, Z.; Luo, Y.; Jiang, X.; Zhang, J.; Li, J.; Zhang, J.; Gong, Y.; Xie, C. PARP inhibitor niraparib as a radiosensitizer promotes antitumor immunity of radiotherapy in EGFR-mutated non-small cell lung cancer. Clin. Transl. Oncol. 2021, 23, 1827–1837.
- Wolchok, J. Putting the Immunologic Brakes on Cancer. Cell 2018, 175, 1452–1454.
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987, 328, 267–270.
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465.
- Pioli, C.; Gatta, L.; Frasca, D.; Doria, G. Cytotoxic T lymphocyte antigen 4 (CTLA-4) inhibits CD28-induced IkappaBalpha degradation and RelA activation. Eur. J. Immunol. 1999, 29, 856–863.
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413.
- Gatta, L.; Calviello, G.; Di Nicuolo, F.; Pace, L.; Ubaldi, V.; Doria, G.; Pioli, C. Cytotoxic T lymphocyte-associated antigen-4 inhibits integrin-mediated stimulation. Immunology 2002, 107, 209–216.
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736.
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895.
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034.
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268.
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297.
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687.
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723.
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86.
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982.
- Huang, Q.; Zheng, Y.; Gao, Z.; Yuan, L.; Sun, Y.; Chen, H. Comparative Efficacy and Safety of PD-1/PD-L1 Inhibitors for Patients with Solid Tumors: A Systematic Review and Bayesian Network Meta-analysis. J. Cancer 2021, 12, 1133–1143.
- Vuky, J.; Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Bellmunt, J.; Powles, T.; Bajorin, D.; Hahn, N.M.; Savage, M.J.; et al. Long-Term Outcomes in KEYNOTE-052: Phase II Study Investigating First-Line Pembrolizumab in Cisplatin-Ineligible Patients With Locally Advanced or Metastatic Urothelial Cancer. J. Clin. Oncol. 2020, 38, 2658–2666.
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060.
- Mullard, A. Second CTLA4-targeted checkpoint inhibitor secures FDA approval. Nat. Rev. Drug Discov. 2022, 21, 868.
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801.
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465.
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986.
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121.
- Nebot-Bral, L.; Brandao, D.; Verlingue, L.; Rouleau, E.; Caron, O.; Despras, E.; El-Dakdouki, Y.; Champiat, S.; Aoufouchi, S.; Leary, A.; et al. Hypermutated tumours in the era of immunotherapy: The paradigm of personalised medicine. Eur. J. Cancer 2017, 84, 290–303.
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Geukes Foppen, M.H.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211.
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199.
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413.
- Abuodeh, Y.; Venkat, P.; Kim, S. Systematic review of case reports on the abscopal effect. Curr. Probl. Cancer 2016, 40, 25–37.
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but Not Single-Dose Radiotherapy Induces an Immune-Mediated Abscopal Effect when Combined with Anti–CTLA-4 Antibody. Clin. Cancer Res. 2009, 15, 5379–5388.
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11, 728–734.
- Rudqvist, N.-P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.-W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; Demaria, S. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol. Res. 2018, 6, 139–150.
- Gong, X.; Li, X.; Jiang, T.; Xie, H.; Zhu, Z.; Zhou, F.; Zhou, C. Combined Radiotherapy and Anti–PD-L1 Antibody Synergistically Enhances Antitumor Effect in Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1085–1097.
- Pilones, K.A.; Hensler, M.; Daviaud, C.; Kraynak, J.; Fucikova, J.; Galluzzi, L.; Demaria, S.; Formenti, S.C. Converging focal radiation and immunotherapy in a preclinical model of triple negative breast cancer: Contribution of VISTA blockade. Oncoimmunology 2020, 9, 1830524.
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys 2013, 86, 343–349.
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74.
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222.
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120.
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758.
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469.
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128.
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218.
- Adams, S.F.; Rixe, O.; Lee, J.-H.; McCance, D.J.; Westgate, S.; Eberhardt, S.C.; Rutledge, T.; Muller, C. Phase I study combining olaparib and tremelimumab for the treatment of women with BRCA-deficient recurrent ovarian cancer. J. Clin. Oncol. 2017, 35, e17052.
- Lee, J.-M.; Cimino-Mathews, A.; Peer, C.J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Cao, L.; Harrell, M.I.; Swisher, E.M.; Houston, N.; et al. Safety and Clinical Activity of the Programmed Death-Ligand 1 Inhibitor Durvalumab in Combination With Poly (ADP-Ribose) Polymerase Inhibitor Olaparib or Vascular Endothelial Growth Factor Receptor 1-3 Inhibitor Cediranib in Women’s Cancers: A Dose-Escalation, Phase I Study. J. Clin. Oncol. 2017, 35, 2193–2202.
- Friedlander, M.; Meniawy, T.; Markman, B.; Mileshkin, L.; Harnett, P.; Millward, M.; Lundy, J.; Freimund, A.; Norris, C.; Mu, S.; et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: Results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 2019, 20, 1306–1315.
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149.
- Lampert, E.J.; Zimmer, A.; Padget, M.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: A Proof-of-Concept Phase II Study. Clin. Cancer Res. 2020, 26, 4268–4279.
- Seyedin, S.N.; Hasibuzzaman, M.M.; Pham, V.; Petronek, M.S.; Callaghan, C.; Kalen, A.L.; Mapuskar, K.A.; Mott, S.L.; Spitz, D.R.; Allen, B.G.; et al. Combination Therapy with Radiation and PARP Inhibition Enhances Responsiveness to Anti-PD-1 Therapy in Colorectal Tumor Models. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 81–92.
- Zhang, N.; Gao, Y.; Huang, Z.; Dai, P.; Luo, Y.; Wu, Q.; Jiang, X.; Sun, W.; Zhang, J.; Han, L.; et al. PARP inhibitor plus radiotherapy reshapes an inflamed tumor microenvironment that sensitizes small cell lung cancer to the anti-PD-1 immunotherapy. Cancer Lett. 2022, 545, 215852.