|
Phases and Timing
|
Main Pathological Manifestations of PBLI
|
|
Phase 1 [2]0–3 h
|
Pulmonary hemorrhage
|
|
|
|
Phase 2 [3]4–24 h
|
Inflammation
|
|
|
|
Phase 3 [4]After 24 h
|
Hypercoagulation
|
|
|
3. Crosstalk among Hemorrhage, Inflammation, and Coagulation
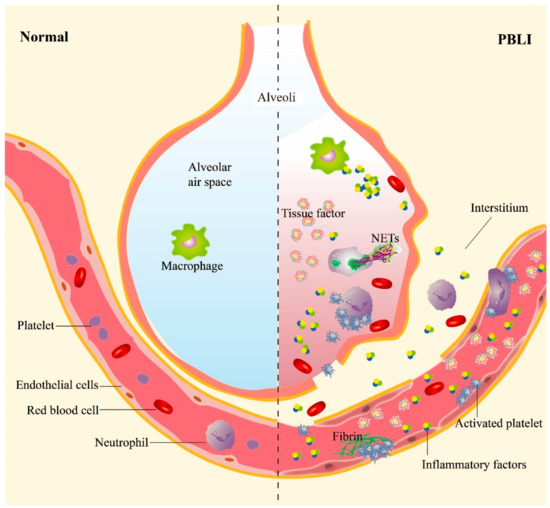
Inflammation and hemorrhage are the two main manifestations of PBLI, subsequently inducing a series of pathological and physiological changes, such as pulmonary edema, alveolar hemorrhage, and emphysema. The processes of inflammation and hemorrhage are not independent but are related to each other (Figure 1). Shockwaves destroy the pulmonary vascular structure directly and induce the activation of platelets, which are essential for maintaining hemostasis following mechanical injury to the vasculature. Meanwhile, inflammatory responses are triggered furtherly.
Figure 1. The main pathological manifestations in PBLI. The main pathological manifestations of PBLI are pulmonary hemorrhage, inflammation, and coagulation disorders. The shockwave caused rupture of the pulmonary capillaries, destruction of the alveoli, and entry of red blood cells into the alveolar space and interstitium. Inflammation is manifested by leukocyte infiltration and increased levels of proinflammatory cytokines in the lung. Coagulation disorders are characterized by the aggregation of activated platelets to form platelet thrombi and the gradual formation of fibrin thrombi. NETs: neutrophil extracellular traps.
3.1. Hemorrhage Promotes Inflammation in PBLI
The existing literature shows that shockwaves could induce platelet activation [
23], expressing a variety of cell surface proteins involved in inflammation. It has been reported that P-selectin, an adhesion molecule present on activated platelets, promotes neutrophil–platelet, platelet–platelet, and monocyte–platelet interactions by binding to P-selectin glycoprotein ligand-1 (PSGL-1) on other cells [
24,
25].
Some studies have shown that soluble CD40L (sCD40L), as a platelet-derived microparticle, shed from the surface of activated platelets, is capable of activating leukocytes and endothelial cells [
26,
27]. Platelet-expressed CD40L interacts with CD40 expressed on endothelial cells, which causes numerous downstream effects, upregulating a number of proinflammatory mediators such as intracellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin [
26].
Studies have confirmed that platelets express low levels of TLRs as pattern recognition receptors in the resting state; once activated, the expression of TLRs is upregulated, which triggers the downstream phosphatidylinositol 3-kinase (PI3K) signaling pathway to activate nuclear factor κB (NF-κB) and promote the release of inflammatory factors (TNF-α, IL-1, and IL-6), chemokines, and adhesion molecules (ICAM-1, VCAM-1, and ELAMs) [
28,
29]. Platelets express various TLRs, among which TLR4 plays a major role in inflammation. TLR4 has been shown to enhance platelet–neutrophil aggregations [
30], and neutrophil extracellular trap (NET) formation in sepsis [
30]. Wu et al. [
31] observed that inhibition of the TLR4 signaling pathway could alleviate the pulmonary inflammatory response in ALI often caused by blunt chest trauma with hemorrhagic shock (THS). Thus, during the process of PBLI, we posit that TLR4 and its ligands play important roles in the post-traumatic immune response and the development of inflammation in PBLI.
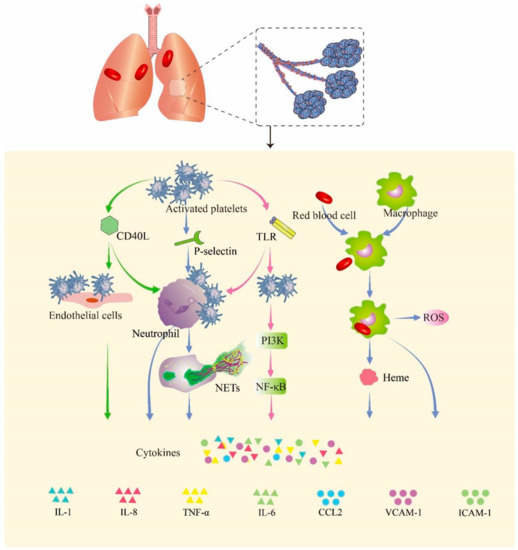
After blast injury, large numbers of hemoglobin-containing red blood cells leak into the alveoli, swallowed by macrophages in the alveoli, and heme is subsequently released from hemoglobin [
32]. Free heme, as a metabolite after the rupture of red blood cells, can induce the proinflammatory response of macrophages. It can induce the production of reactive oxygen species (ROS) and NF-κB signaling molecules in macrophages, thereby promoting the release of inflammatory factors [
33]. In
Figure 2, we summarize the probable pathway of hemorrhage promoting inflammation in PBLI.
Figure 2. Hemorrhage promotes inflammation in PBLI. Activated platelets express a variety of cell surface proteins such as CD40Ls, P-selectins, and TLRs to trigger inflammation responses. The p-selectin expressed by activated platelets mediates the binding of platelets to neutrophils. CD40L mediates the binding of platelets to endothelial cells and neutrophils. The expression of TLRs in platelets promotes binding with neutrophils. Upon binding, neutrophils, endothelial cells, and activated platelets initiate an inflammatory response that promotes the release of cytokines. The red blood cells that leak into the alveoli are swallowed by macrophages and release heme and globulin. Heme induces the release of ROS and activation of the NF-κB signaling pathway in macrophages, thereby triggering inflammation. CD40L: cluster of differentiation 40 ligand; NETs: neutrophil extracellular traps; TLR: Toll-like receptor; PI3K: phosphoinositide 3-kinase; NF-κB: nuclear factor kappa B; ROS: reactive oxygen species; IL-1: interleukin-1; IL-8: interleukin-8; TNF-α: tumor necrosis factor-α; IL-6: interleukin-6; CCL2: C–C motif chemokine ligand 2; VCAM-1: vascular cell adhesion molecule 1; ICAM-1: intercellular adhesion molecule 1.
3.2. Inflammation Aggravates Coagulation Disorders
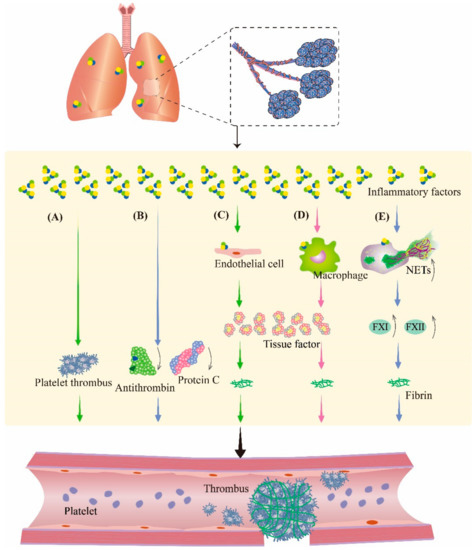
The inflammatory response is a multifactorial defensive process of an organism to injury, such as infectious or noxious stimuli. During inflammation, cytokines modulate the coagulation system; therefore, in the study of inflammation, the involvement of the coagulation pathway must be taken into account. Therefore, it is speculated that inflammation is also crosslinked with coagulation during PBLI (Figure 3). These changes make the management of pulmonary hemorrhage after PBLI more complicated, which deserves further study.
Figure 3. Inflammation aggravates coagulation disorders in PBLI. During inflammation, inflammatory factors can promote thrombosis by regulating the coagulation system and fibrinolytic system. (A) Inflammatory factors promote platelet activation and aggregation to form platelet thrombosis. (B) Inflammatory factors cause the downregulation of antithrombin and protein C to inhibit fibrinolysis. (C,D) Inflammatory factors stimulate endothelial cells and monocytes to produce tissue factor, an activator of coagulation, resulting in fibrinous thrombi. (E) Neutrophils are activated by inflammatory factors to produce NETs, which promote coagulation by promoting the production of several coagulation factors. NETs: neutrophil extracellular traps; FXI: coagulation factor XI; FXII: coagulation factor XII.
Proinflammatory cytokines including interleukins and tumor necrosis factor (TNF) directly promote local coagulation, in addition to being responsible for regulating inflammatory responses [
34]. A previous study [
35] showed that the exposure of whole blood from healthy volunteers to IL-1β, IL-6, and interleukin-8 (IL-8) resulted in hyperactivation of platelets and increased clotting. In addition, TNF promotes platelet aggregation and activation. Several studies also confirmed that proinflammatory cytokines such as TNF-α, IL-1, and IL-6 play important roles in the initiation of coagulation [
36,
37,
38,
39]. In addition to promoting coagulation through the above pathways, proinflammatory cytokines also cause obstacles to the anticoagulant mechanism by inhibiting the activity of antithrombin (AT) and protein C [
38].
TF is expressed on platelets, endotheliocytes, neutrophils, and eosinophils at organ and body surfaces. As the initiator of the extrinsic coagulation pathway, TF plays a central role in inflammation-induced coagulation initiation [
40]. Normally, TF is hardly present in the circulating blood. However, in an inflammatory state, monocytes and endothelial cells can increase the expression of TF through proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 [
36,
41]. The TF expressed by monocytes and neutrophils promotes coagulation in turn.
After activation by cytokines or cytotoxins, neutrophils produce an extracellular fibrous network of DNA, histones, and granulins such as elastase to capture bacteria. This network of extracellular fibers is known as neutrophil extracellular traps (NETs). NETs have been reported to be associated with platelet aggregation and coagulation [
42]. Fuchs et al. [
42] discovered that NETs provide a scaffold and stimulus for platelet binding and aggregation. Histones in NETs or liberated after digestion of NET can also provide a stimulus for platelet aggregation. Cell-free DNA (cfDNA), as a component of NETs, promotes thrombosis by activating some proteases in the coagulation pathway, such as coagulation factors XII and XI, and suppresses fibrinolysis [
43]. In our previous study [
9], we found elevated expression of NETs in the lungs of mice with PBLI, which indicated an increased thrombotic risk in PBLI. After blast exposure, due to the overexpression of proinflammatory factors, TF, and NETs, local coagulation in lung tissue may result, which is contradictory to the pulmonary hemorrhage reaction, further complicating the treatment of PBLI.
4. Pharmacotherapy Principles for PBLI
At present, the clinical treatment of PBLI is mainly based on mechanical ventilation, intensive treatment, and supportive treatment. Pharmacotherapy methods that target different pathological manifestations of PBLI to stop bleeding, inhibit inflammation, and stabilize coagulation may be used to treat PBLI (Table 2).
Table 2. Pharmacotherapies for primary blast lung injury.
|
Drug/Efficacy
|
Drug/Strategy
|
Protective Mechanism
|
Model
|
Author
|
Author
Country
|
Journal Year
|
References
|
|
Hemostasis
|
Recombinant activated factor VII (rFVIIa)
|
Coagulation factor, promotes the production of thrombin
|
BLI patients
|
Martinowitz et al.
|
Israel
|
2004
|
[44]
|
|
Tranexamic acid (TXA)
|
Anti-fibrinolytic agent, impairs fibrinolysis, inhibits clot decomposition
|
Adult trauma patients
|
Roberts
|
UK
|
2015
|
[45]
|
|
Fibrinogen γ-chain-coated adenosine 5′-diphosphate-encapsulated liposomes(H12-(ADP)-liposomes)
|
Targets the injured site, inhibits internal bleeding
|
BLI mice
|
Hagisawa et al.
|
Japan
|
2016
|
[46]
|
|
Thrombin@Fe3O4 nanoparticles
|
Targets the damaged site, promotes the coagulation cascade
|
-
|
-
|
-
|
-
|
-
|
|
Hemostatic dexamethasone nanoparticles (hDNP)
|
Targets the bleeding site, exerts anti-inflammatory effects
|
BLI rats
|
Hubbard et al.
|
US
|
2018
|
[47]
|
|
Anti-inflammation
|
Sivelestat sodium hydrate (sivelestat)
|
Reduces the expression of NE and IL-8
|
Severe burns rats
|
Xiao et al.
|
China
|
2016
|
[48]
|
|
Ulinastatin
|
Reduces the infiltration of inflammatory cells, reduces pulmonary edema and neutrophil infiltration, alleviates lung injury
|
Rats with severe burn–blast combined injury
|
Liu et al.
|
China
|
2018
|
[49]
|
|
BLI rabbits
|
Yuan et al.
|
China
|
2016
|
[50]
|
|
BLI rabbits
|
Dai et al.
|
China
|
2015
|
[51]
|
|
Perfluorocarbon (PFC)
|
Inhibits proinflammatory cytokine release and oxidative stress
|
BLI cells
|
Zhang et al.
|
China
|
2017
|
[52]
|
|
BLI canine
|
Zhang et al.
|
China
|
2020
|
[53]
|
|
N-Acetylcysteine amide (NACA)
|
Decreases myeloperoxidase activity, reduces NF-κB activation, attenuates lung inflammation
|
PBLI rats
|
Chavko et al.
|
US
|
2009
|
[54]
|
|
Anticoagulation
|
Heparin
|
Prevents diffuse intravascular coagulation, improves survival
|
Trauma patients and blast injury rats
|
Yang et al.
|
US
|
2022
|
[55]
|
|
rTFPI
|
Inhibits coagulation cascade
|
Gas explosion rats
|
Tian et al.
|
China
|
2020
|
[56]
|