Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Aging is characterized by a progressive impairment of the physiological functions of tissues and organs. The causes of aging are complex and interconnected, but there is consensus that genomic instability, telomere erosion, epigenetic alteration, and defective proteostasis are primary hallmarks of the aging process.
- senescence-associated secretory phenotype (SASP)
- senescence
- aging
1. Introduction
Aging is defined as the progressive deterioration of an organism, resulting in the progressive impairment of tissue and organ physiological functions [1]. It is characterized by a chronic, sterile, and low-grade inflammation which contributes to the development of age-related diseases, including neurodegeneration, metabolic syndrome, and cardiovascular disorders [2]. The reduction in the ability to manage stress, together with the progressive increase in inflammation that characterizes the aging process has been defined as inflammaging [3]. In this context, a new field of research emerged, called geroscience. It has been developed on the assumption that mechanisms driving aging overlap with those driving age-related diseases, and it tries to unveil the molecular relationship between aging and age-related chronic diseases [4]. To this aim, the trans-NIH Geroscience Interest Group proposed the seven pillars of aging which include stress, macromolecular damage, metabolism, proteostasis, epigenetics, stem cells and regeneration, and inflammation [5]. It is important to underline that such pillars are strongly interconnected and affect each other, thus representing an intricated network that influences aging and age-related diseases.
The seven pillars of aging partially overlap with the nine hallmarks of aging proposed by Lopez-Otin and colleagues in their seminal paper in 2013 [6], to define the causes of age-dependent accumulation in cellular damage [6]. The hallmarks of aging have been subdivided into three main categories: primary, antagonistic, and integrative, which act hierarchically. The primary hallmarks, i.e., nuclear, and mitochondrial genomic instability, telomere erosion, epigenetic alteration, and defective proteostasis, are characterized by their unequivocal participation in the aging process. In fact, any primary hallmark negatively affects cell behavior and leads to aging. On the other hand, antagonistic hallmarks have opposite effects based on their intensity and duration. Antagonistic hallmarks include mitochondrial dysfunction, associated with reactive oxygen species (ROS) generation, deregulated nutrient sensing, and cellular senescence. They initially aim at protecting the cell or the organism from injuries caused by the primary hallmarks but, once chronic, they worsen the damage. Integrative hallmarks, comprising stem cell exhaustion and intercellular communication, directly affect tissue/organism homeostasis. Thus, the antagonistic hallmarks tipping the balance for successful aging should be considered to prevent the insurgence of age-related chronic diseases, as discussed below.
The panel of the research symposium “New Hallmarks of Aging”, held in Copenhagen (Denmark) on the 22nd of March 2022, besides adding new hallmarks, as discussed below, acknowledged that the hallmarks of aging have greater value when viewed as a network rather than individual processes, and that attention to the interconnectedness of different hallmarks should be paid [7].
2. Mitochondrial Dysfunction Associated with ROS Generation
All the primary hallmarks of aging are linked to alteration in the genetic material and its principal metabolic products, proteins, in agreement with the finding that several progeroid syndromes are characterized by DNA damage accumulation [8][9][10].
DNA damage can be induced by endogenous threats such as DNA replication errors and ROS generation [11][12]; both events increase with age and contribute to the insurgence of age-related diseases [13]. DNA damage may also arise from environmental clues, although their specific influences on aging, apart from obvious causes such as smoking and obesity, remain poorly understood [5]. When DNA damage impacts stem cells and compromises their self-renewal properties (an integrative hallmark of aging), tissue functions might be lost [14]. Not only nuclear, but also mitochondrial DNA (mtDNA) is a target for damaging events contributing to the aging process [15]. mtDNA damage can affect mitochondrial fitness by dampening electron transport chain efficiency which, in turn, causes a decrease in ATP production, an accumulation of oxidized NAD+, and an increase in ROS generation. The observed increase in ROS levels with age led to the “free radical theory” of aging proposed by Harman in 1956 [16]. Moreover, ROS also plays a fundamental role in cellular signaling, specifically redox signaling, regulating the functioning of every cell in the organism. Therefore, Sohal and Orr proposed the “redox stress hypothesis” of aging [11], stating that the aging-associated functional decline is primarily driven by the disturbance in redox signaling, i.e., by the imbalance in either oxidative or reductive stress [17]. The concept of redox signaling is one of the reasons to consider mitochondrial dysfunction as an antagonistic hallmark of aging. Low-grade mitochondrial dysfunction, indeed, exerts positive effects on the aging process by lowering the ability to synthesize ATP through oxidative phosphorylation which, in turn, leads to the slowing of the aging process. In fact, as further discussed below, a calorie-restricted diet and intermittent fasting exert beneficial effects on prolonging life span by decreasing metabolic rate and ROS production [18][19].
On the other hand, an increase in ROS production can induce mtDNA damage which has been connected to the activation of the inflammatory responses. In particular, since mitochondrial DNA resembles bacterial genomes, it can be recognized by, and activate, different receptors linked to the immune response, including endosomal TLR9 (toll-like receptor 9) and cytoplasmic inflammasome receptors, which contribute to the onset of inflammatory responses [20]. As an example, mtDNA released by damaged mitochondria activates intracellular inflammasome receptors NLRP3 (NLR family pyrin domain containing 3) and AIM2 (absent in melanoma 2), thus leading to the secretion of the pro-inflammatory cytokine IL-1β and IL-18 [21][22] (see below for further discussion).
In addition to mtDNA damage, an increase in ROS level may induce defective mitophagy, thus dampening the selective removal of malfunctioning mitochondria [15][23]. The ubiquitin-proteasome pathway components E3-ubiquitin ligase Parkin and PTEN-inducible putative kinase 1 (PINK1) are involved in the mitophagic process and recognized as Parkinson's disease (PD)-linked enzymes [24][25].
The increase in ROS generation caused by mitochondrial dysfunction might also affect proteostasis, a primary hallmark of aging [26][27]. The pathogenesis of several age-related diseases has been linked to protein aggregation possibly due to impaired proteostasis, such as the formation of β-amyloid fibrils and neurofibrillary tangles in Alzheimer’s disease, α-synuclein aggregates (Lewy body) in PD, huntingtin aggregates in Huntington’s disease, and islet amyloid polypeptide aggregates in type 2 diabetes [23][28][29][30][31][32][33][34][35]. This could explain why, when mitochondrial dysfunction becomes chronic, it can further increase the rate of the aging process by impairing proteostasis.
The alteration in cellular proteostasis may depend on several factors, including accumulation of aggregated proteins due to DNA mutation or incorrect protein folding, as well as deficits of protein clearance pathways such as ubiquitin-proteasome and autophagy-lysosome pathways, the latter now recognized as a self-standing hallmark of aging [7]. Since these processes are strictly dependent on intracellular ATP levels, it can be postulated that mitochondrial and protein quality control act synergistically to influence age-associated phenotypes.
Moreover, disturbed proteostasis in synergy with inflammation boosts the aging process as demonstrated by the discovery that pro-inflammatory cytokines cause the replacement of the canonical catalytic β subunits in the proteasome complex, leading to the assembly of the immunoproteasome, which selectively degrades proteins involved in inflammation and immune response [36][37]. On the other hand, the inhibition of the immunoproteasome prevents the expression of pro-inflammatory cytokines [38]. Furthermore, increased expression of the immunoproteasome has been demonstrated in AD and amyotrophic lateral sclerosis (ALS) patients, and single nucleotide polymorphisms in immunoproteasome subunits increase the risk of developing neurodegenerative diseases [39]. Thus, mitochondrial dysfunction and the associated ROS generation are deeply interconnected with the other hallmarks of aging and strongly contribute to the fate of an aging cell/organism.
3. Deregulated Nutrient Sensing
As previously mentioned, a calorie-restricted diet and intermittent fasting decrease metabolic rate and ROS production, exerting beneficial effects on the aging process [18][19]. This discovery unequivocally demonstrated that the aging process is strictly dependent on nutrient availability and nutrient-sensing pathways.
Nutrient sensing through insulin/insulin-like growth factor (IGF) was the first pathway proved to regulate the aging process and to be involved in the insurgence of age-related diseases [40][41]. The insulin/IGF-1 signaling has been involved in the aging process by finding that organisms with a constitutively decreased signaling have a longer lifespan, probably because of the decrease in metabolic rate which, as previously discussed, might induce less cellular damage [42]. The insulin/IGF-1 signaling, via the insulin receptor substrate (IRS) adapter, activates phosphoinositide 3-kinase (PI3-K), which, in turn, triggers Akt and mTOR-1 activation. The insulin/IGF1 pathway can be considered an antagonistic hallmark of aging, since reduced mTOR signaling has been associated with longevity, whereas the increased activation of mTOR signaling characterizes the aged phenotype [43]. It is notable that mTOR inhibition lessened mitochondrial dysfunction [44] and protects from genomic instability and telomere attrition in preclinical model organisms [45].
Besides mTOR, AMPK (AMP-activated protein kinase) and sirtuins have also been involved in the aging process through the regulation of nutrient sensing. The decrease in intracellular energy availability can indeed be sensed by both AMPK, which responds to high AMP levels, and sirtuins, histone deacethylases responding to high NAD+ levels. AMPK, for example, can phosphorylate mTOR at Thr2446 leading to its inhibition [46] and thus protecting from age-related diseases. On the other hand, pharmacological, genetic, or stimulus-induced ablation of/reduction in sirtuin activity, leads to premature aging [47][48]. These findings agree with the observed decrease in NAD+ cellular levels during aging [49]. Moreover, nutrient availability controls the function of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of the NAD+ salvage pathway, with calorie restriction increasing its expression [50].
It must also be underlined that sirtuin activity defects, by affecting epigenetic modifications, alter cell epigenetic signature which can be inherited transgenerationally, thus potentially impacting the cell progeny. This is the case for the “trained memory” of innate immune cells which is mediated by epigenetic modification of immune cells (both monocytes and microglia) and has been suggested to produce a long-term hyperinflammatory status sustaining the aging process [48][51].
Besides genome integrity and epigenetic regulation, during the aging process, RNA processing changes also occur. The finding that interventions reversing the senescent phenotype seem to act by restoring splicing factor expression [52] suggested including the dysregulation of RNA processing between the new hallmarks of aging [7].
Furthermore, the aging process also provokes notable changes in the gut microbiome [53], including shifts in microbial populations and loss of species diversity. Together with the age-associated loss of structural integrity of the gut and of the blood–brain barrier these changes in microbial populations can drive inflammation. These observations led to the inclusion of microbiome dysfunction among the new hallmarks of aging [7].
4. Cell Senescence
Primary hallmarks of aging can induce cell senescence, a complex and multifaced process that results in an irreversible proliferation arrest, and resistance to apoptosis. Although senescent cells remain viable, they undergo alterations in metabolic activity [54]. An increase in senescent cell number during the aging process has been reported for several tissues and can reflect both an increase in the rate of senescence and a decrease in senescent cell clearance [6][51][54]. Cell senescence has been acknowledged as an antagonistic hallmark of aging, since it can be a compensatory response to protect the tissue from the proliferation of damaged cells, but if the senescent cell replacement process fails, the accumulation of senescent cells may worsen the damage, thus further contributing to the aging process.
When the senescence program hits stem cells in a tissue, it can cause stem cell exhaustion (integrative hallmark of aging), thus leading to a decline in the regenerative potential culminating in tissue/organ dysfunction [4][6][55]. On the other hand, when the senescence program hits innate and adaptive immune cells, it induces inflammaging, the chronic and sterile inflammation distinctive of the aging process [51][56]. The senescence of immune cells, specifically termed immunosenescence, has been used to construct the inflammatory clock of aging (iAge), a deep-learning method based on patterns of systemic age-related inflammation and immunosenescence. iAge predicts important aging phenotypes and can be utilized for the early detection of age-related clinical manifestations [57].
A key characteristic of senescent cells is the secretion of a specific pattern of molecules collectively known as senescence-associated secretory phenotype (SASP). SASP includes a plethora of soluble signaling factors, such as pro-inflammatory cytokines, chemokines, angiogenic factors, bioactive lipids, and matrix metalloproteinases (MMPs) [58][59][60], which affect the tissue microenvironment potentially driving a self-propelling senescent phenotype.
It has been shown that SASP depends on NAMPT expression and that endothelial SASP impairs insulin signaling in adipocytes [61][62]. These findings corroborate the bidirectional association between cell senescence, dysregulated nutrient sensing, and mitochondrial dysfunction. The pro-inflammatory nature of SASP, the involvement of chronic inflammation in fuelling the self-propelling damaging loop culminating in the insurgence of age-related chronic diseases, and its interconnections with other hallmarks, lead to the recognition of inflammation itself as a new hallmark of aging [7].
One of the pro-inflammatory molecules in SASP is IL-1β, whose secretion requires the inflammasome, a molecular platform that, in response to PAMPs (pathogen-associated molecular patterns) or DAMPs (damage-associated molecular patterns), gets activated and mediates pro-caspase-1 activation via auto-catalytic proteolysis. Active caspase-1, in turn, processes IL-1β that, in its mature form, can be secreted [63][64]. A pivotal role of the inflammasome in aging is demonstrated by the finding that mice lacking the NLRP3 inflammasome receptor have a longer health span and the use of small molecule NLRP3 inhibitors prevents age-related diseases in mice [65]. Moreover, an increase in IL-1β levels in biological fluids has been detected in patients affected by different age-related diseases [66][67][68][69], and inflammasome activation strongly contributes to both inflammaging and immunosenescence [70][71].
SASP contains not only soluble factors but also EVs [54]. These, besides being involved in intercellular communication, have recently gained attention in the context of aging, as their release is altered by aging and, in turn, they strongly impact the aging process (Figure 1).
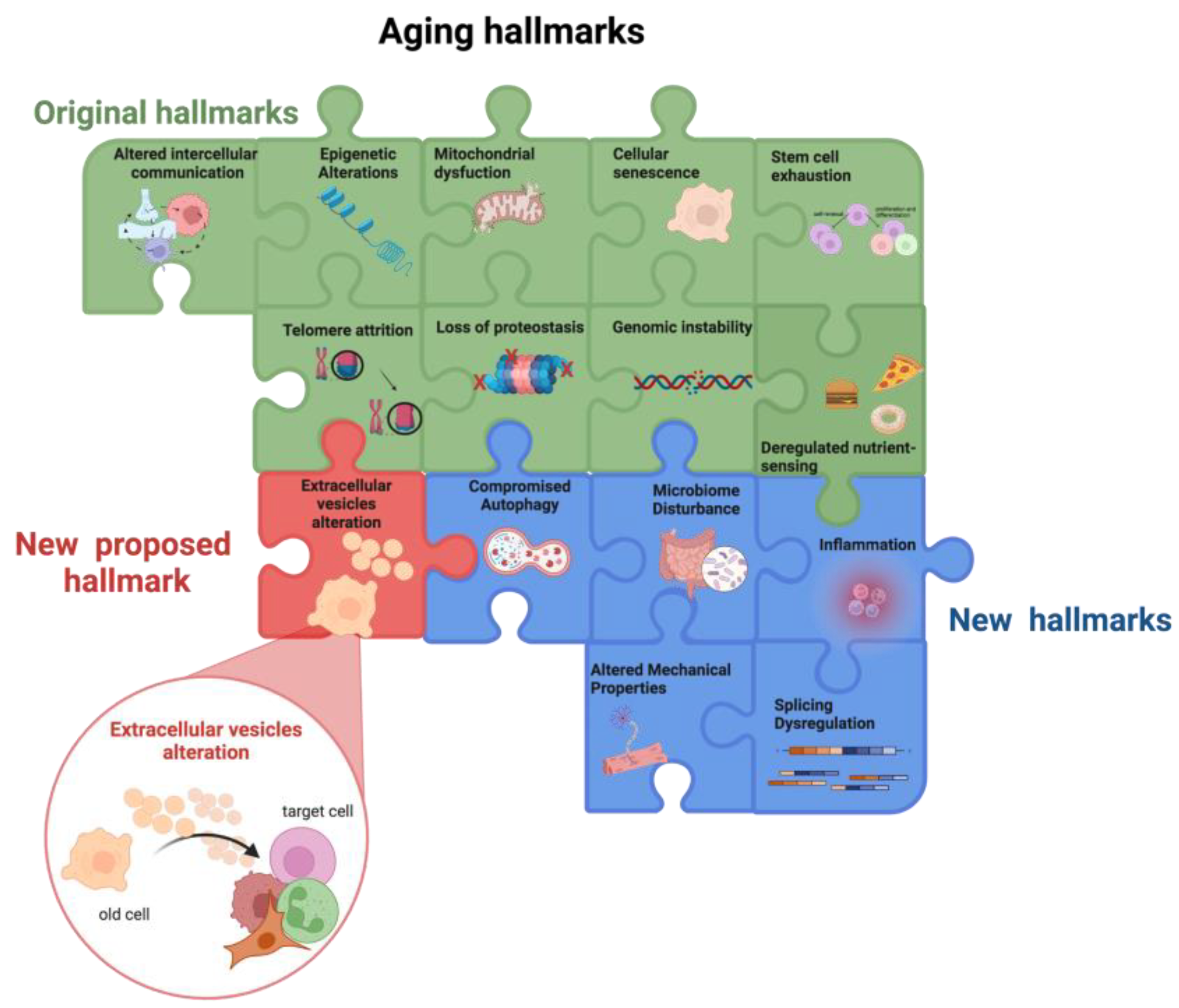
Figure 1. Aging hallmarks. The colored puzzle pieces represent the different hallmarks of aging: in green the original hallmarks and in blue the new hallmarks defined at the Copenhagen Meeting on Aging 2022. Researchers propose “the alteration in extracellular vesicles” as an additional new hallmark of aging (in red). Other potential new hallmarks emerging from future studies will lead to puzzle completion. Created by BioRender.com (accessed on 18 January 2023).
Overall, the main factors involved in the aging process, i.e., the nine hallmarks of aging, influence each other thus creating a vicious cycle that promotes the aging process. Moreover, the bidirectional association between the hallmarks of aging and inflammaging fuels a self-propelling damaging loop culminates in the insurgence of age-related chronic diseases (Figure 1).
This entry is adapted from the peer-reviewed paper 10.3390/cells12040527
References
- Niccoli, T.; Partridge, L. Aging as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752.
- Fulop, T.; Larbi, A.; Pawelec, G.; Khalil, A.; Cohen, A.A.; Hirokawa, K.; Witkowski, J.M.; Franceschi, C. Immunology of Aging: The Birth of Inflammaging. Clin. Rev. Allergy Immunol. 2021, in press.
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254.
- Franceschi, C.; Zaikin, A.; Gordleeva, S.; Ivanchenko, M.; Bonifazi, F.; Storci, G.; Bonafè, M. Inflammaging 2018: An update and a model. Semin. Immunol. 2018, 40, 1–5.
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking aging to chronic disease. Cell 2014, 159, 709–713.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Schmauck-Medina, T.; Molière, A.; Lautrup, S.; Zhang, J.; Chlopicki, S.; Madsen, H.B.; Cao, S.; Soendenbroe, C.; Mansell, E.; Vestergaard, M.B.; et al. New hallmarks of ageing: A 2022 Copenhagen ageing meeting summary. Aging 2022, 14, 6829–6839.
- Spyropoulou, Z.; Papaspyropoulos, A.; Lagopati, N.; Myrianthopoulos, V.; Georgakilas, A.G.; Fousteri, M.; Kotsinas, A.; Gorgoulis, V.G. Cockayne Syndrome Group B (CSB): The Regulatory Framework Governing the Multifunctional Protein and Its Plausible Role in Cancer. Cells 2021, 10, 866.
- Foo, M.X.R.; Ong, P.F.; Dreesen, O. Premature aging syndromes: From patients to mechanism. J. Dermatol. Sci. 2019, 96, 58–65.
- Ashapkin, V.V.; Kutueva, L.I.; Kurchashova, S.Y.; Kireev, I.I. Are There Common Mechanisms between the Hutchinson-Gilford Progeria Syndrome and Natural Aging? Front. Genet. 2019, 10, 455.
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555.
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230.
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772.
- Rossi, D.J.; Jamieson, C.H.; Weissman, I.L. Stems cells and the pathways to aging and cancer. Cell 2008, 132, 681–696.
- Dabravolski, S.A.; Nikiforov, N.G.; Zhuravlev, A.D.; Orekhov, N.A.; Grechko, A.V.; Orekhov, A.N. Role of the mtDNA Mutations and Mitophagy in inflammaging. Int. J. Mol. Sci. 2022, 23, 1323.
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300.
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signalling in oxidative and reductive stress. Biochim. Biophys. Acta 2018, 1865, 721–733.
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99.
- Hatori, M.; Vollmers, C.; Zarrinpar, A.; DiTacchio, L.; Bushong, E.A.; Gill, S.; Leblanc, M.; Chaix, A.; Joens, M.; Fitzpatrick, J.A.; et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012, 15, 848–860.
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 2016, 7, 11–16.
- Pereira, C.A.; Carlos, D.; Ferreira, N.S.; Silva, J.F.; Zanotto, C.Z.; Zamboni, D.S.; Garcia, V.D.; Ventura, D.F.; Silva, J.S.; Tostes, R.C. Mitochondrial DNA Promotes NLRP3 Inflammasome Activation and Contributes to Endothelial Dysfunction and Inflammation in Type 1 Diabetes. Front. Physiol. 2020, 10, 1557.
- Xu, L.; Zhou, J.; Che, J.; Wang, H.; Yang, W.; Zhou, W.; Zhao, H. Mitochondrial DNA enables AIM2 inflammasome activation and hepatocyte pyroptosis in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G1034–G1044.
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150.
- Osman, S. PINK spots: Diseased mitochondria prepare for mitophagy. Nat. Struct. Mol. Biol. 2022, 29, 82.
- Agarwal, S.; Muqit, M.M.K. PTEN-induced kinase 1 (PINK1) and Parkin: Unlocking a mitochondrial quality control pathway linked to Parkinson's disease. Curr. Opin. Neurobiol. 2022, 72, 111–119.
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in aging. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435.
- Rai, M.; Curley, M.; Coleman, Z.; Demontis, F. Contribution of proteases to the hallmarks of aging and to age-related neurodegeneration. Aging Cell 2022, 29, e13603.
- Grottelli, S.; Costanzi, E.; Peirce, M.J.; Minelli, A.; Cellini, B.; Bellezza, I. Potential Influence of Cyclo(His-Pro) on Proteostasis: Impact on Neurodegenerative Diseases. Curr. Prot. Pept. Sci. 2018, 19, 805–812.
- Koyuncu, S.; Saez, I.; Lee, H.J.; Gutierrez-Garcia, R.; Pokrzywa, W.; Fatima, A.; Hoppe, T.; Vilchez, D. The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients. Nat. Commun. 2018, 9, 2886.
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491.
- Henning, R.H.; Brundel, B.J.J.M. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653.
- Kaur, N.; Raja, R.; Ruiz-Velasco, A.; Liu, W. Cellular Protein Quality Control in Diabetic Cardiomyopathy: From Bench to Bedside. Front. Cardiovasc. Med. 2020, 7, 585309.
- Mukherjee, A.; Morales-Scheihing, D.; Butler, P.C.; Soto, C. Type 2 diabetes as a protein misfolding disease. Trends Mol. Med. 2015, 21, 439–449.
- Folger, A.; Wang, Y. The Cytotoxicity and Clearance of Mutant Huntingtin and Other Misfolded Proteins. Cells 2021, 10, 2835.
- Cai, N.; Gomez-Duran, A.; Yonova-Doing, E.; Kundu, K.; Burgess, A.I.; Golder, Z.J.; Calabrese, C.; Bonder, M.J.; Camacho, M.; Lawson, R.A.; et al. Mitochondrial DNA variants modulate N-formylmethionine, proteostasis and risk of late-onset human diseases. Nat. Med. 2021, 27, 1564–1575.
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112.
- Gavilán, M.P.; Castaño, A.; Torres, M.; Portavella, M.; Caballero, C.; Jiménez, S.; García-Martínez, A.; Parrado, J.; Vitorica, J.; Ruano, D. Age-related increase in the immunoproteasome content in rat hippocampus: Molecular and functional aspects. J. Neurochem. 2009, 108, 260–272.
- Ruano, D. Proteostasis Dysfunction in Aged Mammalian Cells. The Stressful Role of Inflammation. Front. Mol. Biosci. 2021, 8, 658742.
- Johnston-Carey, H.K.; Pomatto, L.C.; Davies, K.J. The Immunoproteasome in oxidative stress, aging, and disease. Crit. Rev. Biochem. Mol. Biol. 2015, 51, 268–281.
- Sonntag, W.E.; Lynch, C.D.; Bennett, S.A.; Khan, A.S.; Thornton, P.L.; Cooney, P.T.; Ingram, R.L.; McShane, T.; Brunso-Bechtold, J.K. Alterations in insulin-like growth factor-1 gene and protein expression and type 1 insulin-like growth factor receptors in the brains of ageing rats. Neuroscience 1999, 88, 269–279.
- Sonntag, W.E.; Lynch, C.; Thornton, P.; Khan, A.; Bennett, S.; Ingram, R. The effects of growth hormone and IGF-1 deficiency on cerebrovascular and brain ageing. J. Anat. 2000, 197, 575–585.
- van Heemst, D. Insulin, IGF-1 and longevity. Aging Dis. 2010, 1, 147–157.
- Mota-Martorell, N.; Jové, M.; Pamplona, R. mTOR Complex 1 Content and Regulation Is Adapted to Animal Longevity. Int. J. Mol. Sci. 2022, 23, 8747.
- Walters, H.E.; Deneka-Hannemann, S.; Cox, L.S. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging 2016, 8, 231–244.
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395.
- Cheng, S.W.; Fryer, L.G.; Carling, D.; Shepherd, P.R. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J. Biol. Chem. 2004, 279, 15719–15722.
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell Cardiol. 2007, 43, 571–579.
- Fanucchi, S.; Dominguez-Andres, J.; Joosten, L.A.B.; Netea, M.G.; Mhlanga, M.M. The Intersection of Epigenetics and Metabolism in Trained Immunity. Immunity 2021, 54, 32–43.
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; de Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546.
- Song, J.; Ke, S.F.; Zhou, C.C.; Zhang, S.L.; Guan, Y.F.; Xu, T.Y.; Sheng, C.Q.; Wang, P.; Miao, C.Y. Nicotinamide phosphoribosyltransferase is required for the calorie restriction-mediated improvements in oxidative stress, mitochondrial biogenesis, and metabolic adaptation. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 44–57.
- Teissier, T.; Boulanger, E.; Cox, L.S. Interconnections between inflammaging and immunosenescence during aging. Cells 2022, 11, 359.
- Latorre, E.; Harries, L.W. Splicing regulatory factors, ageing and age-related disease. Ageing Res. Rev. 2017, 36, 165–170.
- Wilmanski, T.; Diener, C.; Rappaport, N.; Patwardhan, S.; Wiedrick, J.; Lapidus, J.; Earls, J.C.; Zimmer, A.; Glusman, G.; Robinson, M.; et al. Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat. Metab. 2021, 3, 274–286.
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell. Dev. Biol. 2021, 9, 645593.
- Ahmed, A.S.; Sheng, M.H.; Wasnik, S.; Baylink, D.J.; Lau, K.W. Effect of aging on stem cells. World J. Exp. Med. 2017, 7, 1–10.
- Mittelbrunn, M.; Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 2021, 22, 687–698.
- Sayed, N.; Huang, Y.; Nguyen, K.; Krejciova-Rajaniemi, Z.; Grawe, A.P.; Gao, T.; Tibshirani, R.; Hastie, T.; Alpert, A.; Cui, L.; et al. An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat. Aging 2021, 1, 598–615.
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118.
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868.
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576.
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 2019, 21, 397–407.
- Barinda, A.J.; Ikeda, K.; Nugroho, D.B.; Wardhana, D.A.; Sasaki, N.; Honda, S.; Urata, R.; Matoba, S.; Hirata, K.I.; Emoto, N. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat. Commun. 2020, 11, 481.
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36.
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Ann. Rev. Immunol. 2009, 27, 519–550.
- Lee, K.A.; Robbins, P.D.; Camell, C.D. Intersection of immunometabolism and immunosenescence during aging. Curr. Opin. Pharmacol. 2021, 57, 107–116.
- Ikonomidis, I.; Andreotti, F.; Economou, E.; Stefanadis, C.; Toutouzas, P.; Nihoyannopoulos, P. Increased proinflammatory cytokines in patients with chronic stable angina and their reduction by aspirin. Circulation 1999, 100, 793–798.
- Yndestad, A.; Damås, J.K.; Oie, E.; Ueland, T.; Gullestad, L.; Aukrust, P. Systemic inflammation in heart failure-the whys and wherefores. Heart Fail Rev. 2006, 11, 83–92.
- Seppi, D.; Puthenparampil, M.; Federle, L.; Ruggero, S.; Toffanin, E.; Rinaldi, F.; Perini, P.; Gallo, P. Cerebrospinal fluid IL-1β correlates with cortical pathology load in multiple sclerosis at clinical onset. J. Neuroimmunol. 2014, 270, 56–60.
- Mendiola, A.S.; Cardona, A.E. The IL-1β phenomena in neuroinflammatory diseases. J. Neural. Transm. 2018, 125, 781–795.
- Brahadeeswaran, S.; Sivagurunathan, N.; Calivarathan, L. Inflammasome Signalling in the Aging Brain and Age-Related Neurodegenerative Diseases. Mol. Neurobiol. 2022, 59, 2288–2304.
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795.
This entry is offline, you can click here to edit this entry!