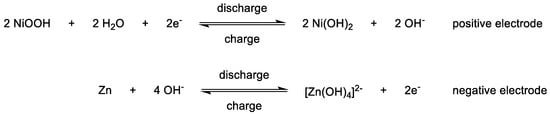
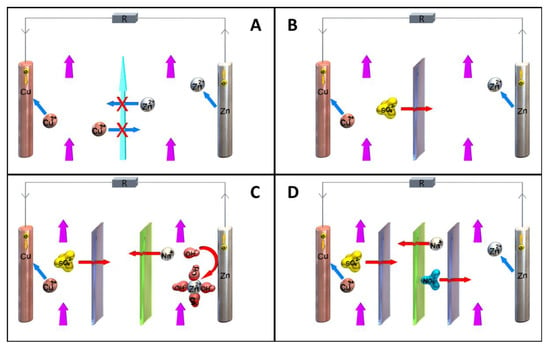
A newer development in RFBs, due to their increasing complexity and compatibility requirements, is the creation of bipolar electrolytes, i.e., electrolytes containing a catholyte and an anolyte function simultaneously [
159]. Although this generally requires a larger synthetic effort and imbues the active electrolyte with one to be solubilized component that is inactive at all times, the major advantage is that it eliminates capacity loss from cross-over [
106,
159]. An oxidized bipolar electrolyte traversing the membrane will regain its electron, reducing energy efficiency, but will then go on to take an active part in the charging process on the other side, and vice versa. One key requirement for favorable energy density in bipolar electrolytes is that the redox events are substantially separated. The presence of two successive redox events alone, such as in viologens (
A5), does not compete with state of the art when the OCP is only 0.5 V. Notably, conditions as subtle as the selected positive counterion in the supporting electrolyte can contribute significantly to the separation, as shown in an analogous pyridinium electrolyte [
160], ultimately having a much more crucial impact on bipolar activity than on two-electron anolyte or catholyte storage. The poor compatibility of Li
+ towards retaining redox potentials was noted in the abovementioned work of Zhang et al. on azobenzene also [
103], although Li
+-coordination with the charged anolyte state had a stabilizing effect. On the other hand, this compensatory effect was found to be absent for Wei and coworkers, with Li
+, specifically out of a range of supporting electrolytes, having a substantial destabilizing effect on the fluorenone anolyte despite functioning well on the catholyte side [
117].
2.5. Summary
Granted that the make-up of the RFB contains many complexly interrelated facets and that any given system may respond differently to minor changes, the trends observed above may contribute strongly to improving overall battery performance:
-
Acetonitrile is the solvent of choice, given its low viscosity and toxicity, and importantly, its polar nature, which generally contributes to solubilizing charged states also.
-
Generally, TFSI− and TEA+ are preferred supporting ions. Alternatively, TBAPF6 is also often applied and performs well. Li+, notably, has exhibited some incompatibility issues in the anolyte compartment.
-
Electron-withdrawing substituents on catholytes and electron donating substituents on anolytes can slightly increase the system’s voltage, though a possible negative effect on stability may negate the gain in OCV.
-
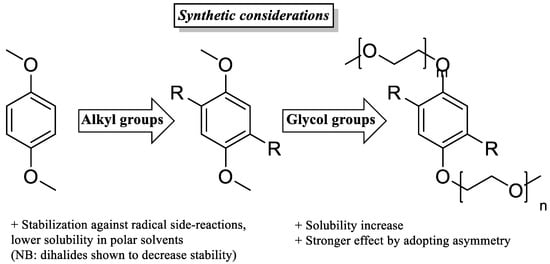
Appending ethylene glycol (preferably asymmetrically) or tetraalkylammonium chains substantially increases the solubility in acetonitrile and to date seems to be the most efficient way to non-invasively increase energy density.
-
Imbuing both the catholyte and anolyte with permanent charge, for example, with tetraalkylammonium side-groups, enables the formation of electrolyte salts. This approach is especially promising, as it discards the need for a supporting electrolyte, since solely the counter-ion (e.g., TFSI) needs to traverse the anion exchange membrane, thereby simultaneously maximizing the concentration of active species and combating capacity decay from cross-over of electrolyte.
3. Introducing a RAS from Basic Lab-Scale Identification towards in-Flow RFB Measurements
3.1. Main RFB-Cell Components
3.1.1. Reference Electrode
The implementation of reference electrodes enables monitoring of either the anolyte or the catholyte compartment during galvanostatic battery charge and discharge (see
Section 4.2.) [
165,
166,
167,
168]. With this, informative polarization techniques can be made, as they touch the limits of any redox electrolyte [
169]. Ultimately, measuring an accurate cell voltage is highly important to guarantee a safe and reversible electrochemical reaction, as many efficiency-lowering processes are due to overpotential-induced decomposition side reactions. Therefore, the reference electrode has to be placed in the opposite half-cell of the current-applying electrode.
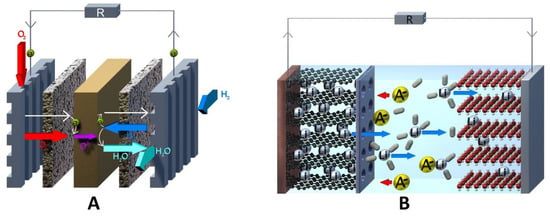
3.1.2. Current Collector, Flow Field, and Bipolar Plate
The differentiation within a half-cell between current collector, bipolar plate, and flow field can be misunderstood, since the respective component often fulfills multiple and mutually substitutive functions. The current collector works as an electrical connection to the circuit outside and reaction surface, and the flow field acts as a turbulent inducer to ensure good electrolyte distribution over the current collector’s entire surface [
13]. The bipolar plate combines these properties, as it plays both a fluidic and an electrodynamic role, serving as a reaction surface and turbulence inducer [
170]. For this, several different technical approaches have been developed, e.g., turbulent flow-through electrodes [
171], “piece of pie” cell configuration [
172], static mixer electrodes [
173], and zero-gap cell designs [
18,
174]. However, the majority of research-scale RFB cell setups operate with the zero-gap approach. Here, all cell materials (i.e., conductive ground plates, carbon felts, and membrane) used are compressed by the cell to minimize distance-induced resistances. In a zero-gap configuration, conductive ground plates play a dual hydro- and electrodynamic role. The embedded flow frame guides redox electrolytes within an inner compartment of a flow cell, ultimately boosting the conversion rate and preventing an overpotential build-up over the entire electroactive surface [
175]. Notably, this strategy has been subject to many computational studies over the past years [
16,
176,
177]. On the other side, conductive plates act as current collectors, securing an electrical interfacing with external energy sources or loads [
178]. To alter a diffusion regime, to extend an accessible electrochemically active area, and therefore, to ensure more effective mass conversion on the surface of current collector, a macro-porous 3D block of compressible and conductive material is normally introduced on top of the ground plate [
179,
180]. This configuration enables a considerably improved range of operational current densities supported by RFBs, although the range of accessible electrolyte flow rates is slightly decreased due to the pressure drop over an additional physical obstacle [
181,
182]. Typical choices for porous current collectors are carbon-based materials—thermally altered, chemically altered, or pristine versions of carbon/graphite felts [
183,
184,
185] or carbon/graphite paper [
186], and in some cases, well-defined reticulated vitreous carbon foams [
187].
3.1.3. Separator
A separator, sometimes called “the heart of an RFB”, effectively separates the anolyte and the catholyte compartment electrolytes and serves to selectively permeate certain species of redox electrolytes [
188,
189,
190]. Separators are conventionally classified referring to their transport mechanisms: (cation/anion/bipolar) selective-ion exchange membranes, microporous size-exclusion separators, ion-conductive glass ceramics, or an interface of immiscible redox electrolytes. In the context of RFBs, the term “membrane” refers to solid polymer ion-selective membranes, whereas a separator uses size exclusion [
34,
191]. Typical cation exchange membranes (CEM) consist of fluorocarbon- or hydrocarbon-based polymers with embedded terminal sulfonic groups, providing a rigid hydrophobic exterior and hydrophilic pore, where the cationic transport occurs. In turn, typical anion exchange membranes (AEM) incorporate terminal tetraalkylammonium groups. The special case, lastly, is bipolar exchange membranes, which are multilayer combinations of anion and cation conductive polymers.
3.2. Practical Difficulties of RAS Transversion and Their Theoretical Background
Introducing a new RAS into the area of redox flow brings about several new challenges. Compared to first fundamental investigations of the electrochemical behavior in a classical three-electrode setup, transferring to a RFB setup (i) changes electrode geometry and size, (ii) adds a separator between electrodes, and (iii) adds fluid-related mechanical effects, such as viscosity. The concurrent introduction of fluid mechanics during in-flow RFB cycling intensifies additional effects of mass transport and iR drop, leading to further changes in electron transfer kinetics during cycling. Simplified, these effects can be summarized in additional overpotentials in electrochemical measurements. As overpotentials have a huge impact on the reversibility, stability, and durability on long-term battery cycling, it is important to identify these. For this, an easy and fast technique such as cyclic voltammetry (CV) can be used.
3.2.1. Cell Setup and Processing Impacts on Mass Transport, Conversion, and Overpotential
In initial proof-of-concept electrochemical measurements, a classic three-electrode CV measurement should be performed to identify the RAS redox potential under “ideal” conditions. Additionally, this stage should be used to identify further important RAS specific characteristics, such as reversibility (basically peak-to-peak separation), diffusional coefficient
D0, standard rate constant
k0, or charge-transfer coefficient
α [
197,
198].
When changing from proof-of-concept electrochemical to static RFB measurements, two main points change: the electrode surface size, and often also material, and the introduction of a separator between the working electrode and the counter and reference electrode. In terms of the electrode surface area for a classical disc macro-electrode, a linear semi-infinite diffusion model is assumed, where the peak-to-peak separation for reversible systems is equal to 2.218
RT/F [
199]. However, if a macroscopically structured electrode material (e.g., carbon nanotubes, carbon foams, or graphite plates) is studied, the diffusion model changes. In summary, these enhancements in mass transport reduce the previously explained overpotential-induced peak-to-peak separation and further increase the peak intensities. Since for in-flow potentiodynamic measurements, the diffusive mass transport at the electrode surface is now superimposed by convection, the peak-limiting current of the CV measurement no longer is achieved by mass transport but by high SOC. The scan rate must be selected so low that the volume current ensures complete circulation of the electrolyte within 59 mV. As a higher SOC must be achieved for these measurements, both the stability of the RAS and the prevention of crossover of the RAS can be tested easily and quickly.
3.2.2. Membrane’s Impacts on Mass Transport, Conversion, and Overpotential
In order for the anolyte and catholyte compartment reactions to take place independently, a separator needs to be used. A semipermeable membrane, ideally, allows for passage of only the charge balancing ions, and not the RAS themselves [
204]. To achieve this, one of two different mechanisms are applied: (i) charge exclusion or (ii) size exclusion [
205,
206]. In both cases, the separator is, in the electrochemical sense, an additional resistor in the system, as the crossing of charge-balancing species requires a certain activation energy [
189]. For size exclusion, this mainly originates from the limited mass transport in the direct area of the membrane surface by Fickian’s law. For charge exclusion, specifically, coulombic repulsion additionally takes place, so that overall the introduced resistance is higher than that of a size exclusion separator [
146,
206]. For multiple separator setups, this resistance increases with every additional separator.
3.2.3. Viscosity’s Impacts on Mass Transport, Conversion, and Overpotential
In addition to the electrochemical changes described in the previous section, fluid mechanics play a large role in energy efficiency in-flow, largely effected by the porosity of the electrode and viscosity of the electrolyte solution [
209,
210]. A distinction can be made between the viscosity influence at the macroscopic level (I) and the atomic level (II). At the macroscopic level (I), viscosity acts like a force working against the flowing electrolyte, demanding more pump energy to maintain a constant volume flow with increasing viscosities [
211]. Simplified, this can be described for a homogeneous Newtonian fluid in a straight pipe by the
Hagen–poiseuille equation, with the volume flow being inversely proportional to the viscosity.
3.3. Summary
While moving towards in-flow RFB measurements, a phased approach of introducing the RAS to the RFB allows a stepwise identification of effects such as diffusional and convectional mass transport, Coulombic repulsion, and fluid mechanical changes, such as viscosity. This causes overpotentials and an iR drop. These effects are highly dependent on electrolyte composition and RFB setup and therefore, the optimum varies from RFB to RFB (electrolyte composition, type of membrane, flow rate, current density, type and geometry of electrode). For this, the step-by-step transferring sequence to better understand the transitioning effects and their intensity is emphasized: (1) proof-of-concept electrochemical measurements with classic CV three-electrode setup; (2) stationary RFB CV measurements; (3) in-flow RFB CV measurements. In the first part, the basic functionality of a RAS should be validated (i.e., redox potential, reversibility). In the second step, the impacts of RFB cell, electrode type, and membrane on the results obtained in the first part are evaluated. Finally, in the third step, the impact of volume flow is investigated, along with the minimization of cross-over. The results of this step-by-step sequence can then be used to define optimum charge-discharge parameters and cut-off criteria in order to obtain the most stable, safe, and durable RFB settings.