Alzheimer’s disease (AD) is a devastating and irreversible neurodegenerative disorder with unknown etiology. While its cause is unclear, a number of theories have been proposed to explain the pathogenesis of AD. In large part, these have centered around potential causes for intracerebral accumulation of beta-amyloid (βA) and tau aggregates. Yet, persons with AD dementia often exhibit autopsy evidence of mixed brain pathologies including a myriad of vascular changes, vascular brain injuries, complex brain inflammation, and mixed protein inclusions in addition to hallmark neuropathologic lesions of AD, namely insoluble βA plaques and neurofibrillary tangles (NFTs). Epidemiological data demonstrate that overlapping lesions diminish the βA plaque and NFT threshold necessary to precipitate clinical dementia. Moreover, a subset of persons who exhibit AD pathology remain resilient to disease while other persons with clinically-defined AD dementia do not exhibit AD-defining neuropathologic lesions. It is increasingly recognized that AD is a pathologically heterogeneous and biologically multifactorial disease with uncharacterized biologic phenomena involved in its genesis and progression.
- Alzheimer’s disease
- beta-amyloid (βA)
- glymphatic–lymphatic system
- immune cell
- macrophage
- synapse
- tau
1. Introduction
1.1. Neuropathologic Criteria for AD
1.2. Theories on AD Pathogenesis
2. Discordance between Neuropathologic Grading Schemes and Theories of AD
Despite the requirement of βA plaques and NFTs for diagnosis of AD-NC and the use of βA as a surrogate endpoint of disease, these markers are of limited utility in disease grading. For instance, precise form(s) and toxic βA species are ambiguous and associations of DP density with cognitive impairment are weak. On the other hand, the abundance and distribution of NFTs and NPs are thought to be predictors for cognitive impairment [2][14]. Yet, NFTs and NPs are also commonly observed in individuals who are cognitively intact [14]. An investigation into risk factors associated with rapid clinical progression, from MCI to dementia, highlights the significance of cerebrospinal fluid (CSF) total tau and phosphorylated tau, but not βA1–42 levels, as pertinent to disease [15]. Moreover, abnormal tau protein inclusions also arise in association with other neurodegenerative conditions (i.e., tauopathies), including Pick disease, progressive supranuclear palsy, corticobasal degeneration, chronic traumatic encephalopathy, aging-related tau astrogliopathy, primary age-related tauopathy, and some forms of frontotemporal dementia [16]. Thus, NFTs and NPs are nonspecific lesions that do not accurately predict elderly persons with AD dementia [17][18]. Meanwhile, a number of alternate brain changes and protein inclusions not encompassed by AD-NC criteria are commonly associated with AD. While AD-NC, vascular diseases, and brain vascular injuries are recognized as the most common neuropathologic lesions associated with AD dementia, these entities also often overlap [19]. For a variety of reasons, it has increasingly been thought that βA is associated with, but not causative of AD. It is also increasingly accepted that significant subsets of individuals with AD dementia exhibit heterogeneous tissue substrates and unique combinations of tissue lesions [20].
Shifting Perspectives: Inconsistencies of the Amyloid Cascade Hypothesis
3. New Theories: Convergence on Vascular and Neuroimmune Homeostatic Factors
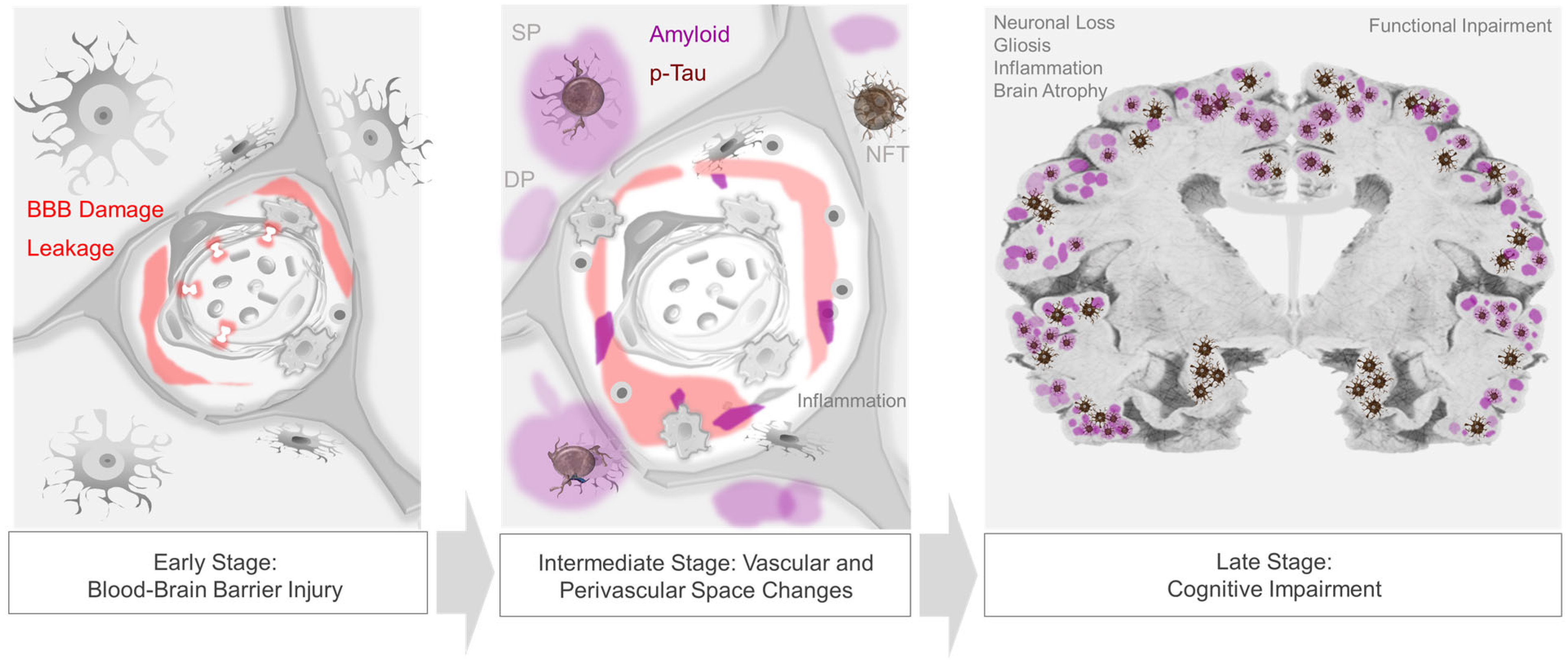
Over the past two decades, investigation into alternative and early-stage AD biomarkers has underscored potential new AD targets [7][30]. Given the convergence of a number of early biological phenomena around cerebral vessels, it is now clear that cerebrovascular contributions to AD have been underrecognized. Increasing data show that inflammatory changes and loss of vascular wall integrity are the earliest identifiable lesions in persons with AD, preceding βA and tau accumulation [31][32].
In parallel, improvements in microscopy and imaging technologies have led to revised concepts in neuroanatomy, neuroimmunology, and brain barrier biology. The meningeal lymphatic system has emerged as a critical player in neurophysiology [33][34][35].
3.1. Brain Border Macrophages in AD
3.2. Peripheral Monocytes and Macrophage Infiltration in AD
3.3. Microgliosis and Other Vascular-Immune Factors in AD
3.4. Imaging Evidence of Early Hypometabolism and Vascular and Perivascular Dysfunction
3.5. Cardiac and Cardiovascular Disease Effects in AD
4. Potential Roles of the Glymphatic–Lymphatic System in AD
4.1. Clearance Mechanisms at the Neurovascular-Perivascular Interface
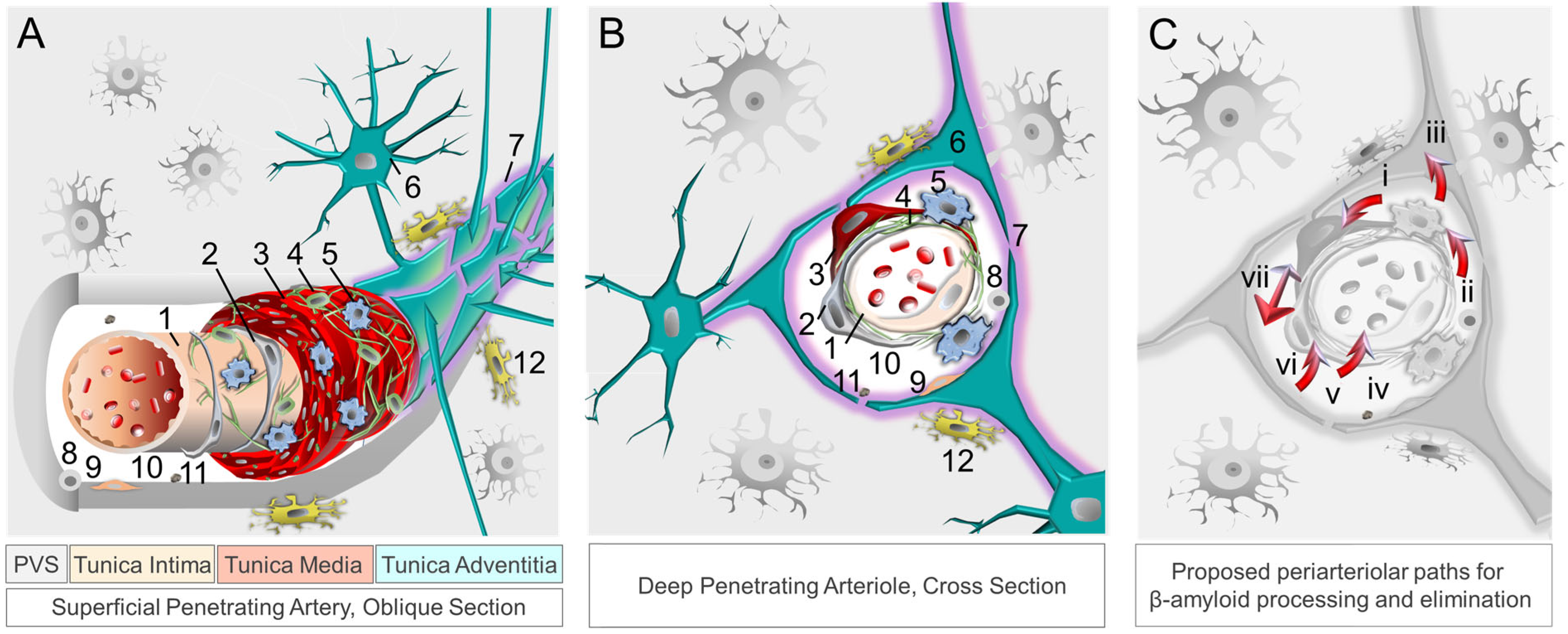
4.2. Compound Proteinopathies in AD
As highlighted in recent literature, significant subsets of AD dementia cases are attributable to TDP-43 inclusions and/or α-synuclein-positive Lewy bodies [16][88][89]. Studies show that non-βA and non-tau proteinopathies are not mutually exclusive of one another, nor with AD-NC [88][89]. In fact, cohort studies demonstrate that TDP-43 pathology combined with AD-NC accounts for 35–37% of pathology in elderly subjects with AD dementia [90][91], while Lewy body inclusions overall manifest in more than 50% [91][92]. Thus, common mechanisms may exist for proteinopathic diseases [16][93].
4.3. Blood, Peripheral Signaling and Brain-Body Connections in AD
Intracranial vessels (i.e., lymphatic and blood vascular) and biofluids (i.e., ISF, CSF, and blood) convey innumerable signals. Secretory mechanisms at the BBB impart endocrine-like properties of BBB endothelium [94][95]. Moreover, neurons and neuroglial cells communicate with themselves, with one another, and with the periphery via receptors and extracellular molecules involving autocrine, paracrine, and endocrine signals that transmit in extracellular spaces [94][95][96]. Other serum-derived factors and biomolecules derived from brain and periphery are also transported through intracranial fluids [97][98][99][100][101][102][103].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11020408
References
- Hyman, B.T.; Trojanowski, J.Q. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 1997, 56, 1095–1097.
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8, 1–13.
- Kumar, A.; Nemeroff, C.B.; Cooper, J.J.; Widge, A.; Rodriguez, C.; Carpenter, L.; McDonald, W.M. Amyloid and Tau in Alzheimer’s Disease: Biomarkers or Molecular Targets for Therapy? Are We Shooting the Messenger? Am. J. Psychiatry 2021, 178, 1014–1025.
- Dickson, T.C.; Vickers, J.C. The morphological phenotype of beta-amyloid plaques and associated neuritic changes in Alzheimer’s disease. Neuroscience 2001, 105, 99–107.
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target Ther. 2019, 4, 29.
- Das, B.; Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263.
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608.
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 2021, 17, 115–124.
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128.
- Woods, J.; Snape, M.; Smith, M.A. The cell cycle hypothesis of Alzheimer’s disease: Suggestions for drug development. Biochim. Biophys. Acta 2007, 1772, 503–508.
- Arshavsky, Y.I. Alzheimer’s Disease: From Amyloid to Autoimmune Hypothesis. Neuroscientist 2020, 26, 455–470.
- Penner, M.R.; Roth, T.L.; Barnes, C.A.; Sweatt, J.D. An epigenetic hypothesis of aging-related cognitive dysfunction. Front. Aging Neurosci. 2010, 2, 9.
- Lemke, G.; Huang, Y. The dense-core plaques of Alzheimer’s disease are granulomas. J. Exp. Med. 2022, 219, e20212477.
- Crystal, H.; Dickson, D.; Fuld, P.; Masur, D.; Scott, R.; Mehler, M.; Masdeu, J.; Kawas, C.; Aronson, M.; Wolfson, L. Clinico-pathologic studies in dementia: Nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology 1988, 38, 1682–1687.
- Van Rossum, I.A.; Visser, P.J.; Knol, D.L.; van der Flier, W.M.; Teunissen, C.E.; Barkhof, F.; Blankenstein, M.A.; Scheltens, P. Injury markers but not amyloid markers are associated with rapid progression from mild cognitive impairment to dementia in Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 319–327.
- Mehta, R.I.; Schneider, J.A. Neuropathology of the Common Forms of Dementia. Clin. Geriatr. Med. 2023, 39, 91–107.
- Gao, Y.L.; Wang, N.; Sun, F.R.; Cao, X.P.; Zhang, W.; Yu, J.T. Tau in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175.
- Ryder, B.D.; Wydorski, P.M.; Hou, Z.; Joachimiak, L.A. Chaperoning shape-shifting tau in disease. Trends Biochem. Sci. 2022, 47, 301–313.
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562.
- Kawas, C.H.; Corrada, M.M. Successful cognitive aging: What the oldest-old can teach us about resistance and resilience. Neurology 2020, 95, 329–330.
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118.
- Carroll, T.; Guha, S.; Nehrke, K.; Johnson, G.V.W. Tau Post-Translational Modifications: Potentiators of Selective Vulnerability in Sporadic Alzheimer’s Disease. Biology 2021, 10, 1047.
- Zhou, B.; Lu, J.G.; Siddu, A.; Wernig, M.; Südhof, T.C. Synaptogenic effect of APP-Swedish mutation in familial Alzheimer’s disease. Sci. Transl. Med. 2022, 14, eabn9380.
- Condello, C.; Merz, G.E.; Aoyagi, A.; DeGrado, W.F.; Prusiner, S.B. βA and Tau Prions Causing Alzheimer’s Disease. Methods Mol. Biol. 2023, 2561, 293–337.
- Boyle, P.A.; Wilson, R.S.; Yu, L.; Barr, A.M.; Honer, W.G.; Schneider, J.A.; Bennett, D.A. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann. Neurol. 2013, 74, 478–489.
- Satizabal, C.L.; Beiser, A.S.; Chouraki, V.; Chêne, G.; Dufouil, C.; Seshadri, S. Incidence of Dementia over Three Decades in the Framingham Heart Study. N. Engl. J. Med. 2016, 374, 523–532.
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683.
- Boyle, P.A.; Yang, J.; Yu, L.; Leurgans, S.E.; Capuano, A.W.; Schneider, J.A.; Wilson, R.S.; Bennett, D.A. Varied effects of age-related neuropathologies on the trajectory of late life cognitive decline. Brain 2017, 140, 804–812.
- Boyle, P.A.; Yu, L.; Wilson, R.S.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann. Neurol. 2018, 83, 74–83.
- Haage, V.; De Jager, P.L. Neuroimmune contributions to Alzheimer’s disease: A focus on human data. Mol. Psychiatry 2022, 27, 3164–3181.
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276.
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302.
- Da Mesquita, S.; Fu, Z.; Kipnis, J. The Meningeal Lymphatic System: A New Player in Neurophysiology. Neuro 2018, 100, 375–388.
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341.
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999.
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235.
- Kierdorf, K.; Masuda, T.; Jordão, M.J.C.; Prinz, M. Macrophages at CNS interfaces: Ontogeny and function in health and disease. Nat. Rev. Neurosci. 2019, 20, 547–562.
- Drieu, A.; Du, S.; Storck, S.E.; Rustenhoven, J.; Papadopoulos, Z.; Dykstra, T.; Zhong, F.; Kim, K.; Blackburn, S.; Mamuladze, T.; et al. Parenchymal border macrophages regulate the flow dynamics of the cerebrospinal fluid. Nature 2022, 611, 585–593.
- Mestre, H.; Verma, N.; Greene, T.D.; Lin, L.A.; Ladron-de-Guevara, A.; Sweeney, A.M.; Liu, G.; Thomas, V.K.; Galloway, C.A.; de Mesy Bentley, K.L.; et al. Periarteriolar spaces modulate cerebrospinal fluid transport into brain and demonstrate altered morphology in aging and Alzheimer’s disease. Nat. Commun. 2022, 13, 3897.
- Hawkes, C.A.; McLaurin, J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266.
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472.
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438.
- Malm, T.M.; Koistinaho, M.; Pärepalo, M.; Vatanen, T.; Ooka, A.; Karlsson, S.; Koistinaho, J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol. Dis. 2005, 18, 134–142.
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502.
- Stalder, A.K.; Ermini, F.; Bondolfi, L.; Krenger, W.; Burbach, G.J.; Deller, T.; Coomaraswamy, J.; Staufenbiel, M.; Landmann, R.; Jucker, M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J. Neurosci. 2005, 25, 11125–11132.
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43.
- Gao, C.; Shen, X.; Tan, Y.; Chen, S. Pathogenesis, therapeutic strategies and biomarker development based on “omics” analysis related to microglia in Alzheimer’s disease. J. Neuroinflamm. 2022, 19, 215.
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675.
- Pillai, J.A.; Maxwell, S.; Bena, J.; Bekris, L.M.; Rao, S.M.; Chance, M.; Lamb, B.T.; Leverenz, J.B.; Alzheimer’s Disease Neuroimaging Initiative. Key inflammatory pathway activations in the MCI stage of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2019, 6, 1248–1262.
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015, 6, 6176.
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803.
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346.
- Welikovitch, L.A.; Carmo, S.D.; Maglóczky, Z.; Malcolm, J.C.; Lőke, J.; Klein, W.L.; Freund, T.; Cuello, A.C. Early intraneuronal amyloid triggers neuron-derived inflammatory signaling in APP transgenic rats and human brain. Proc. Natl. Acad. Sci. USA 2020, 117, 6844–6854.
- Herber, D.L.; Mercer, M.; Roth, L.M.; Symmonds, K.; Maloney, J.; Wilson, N.; Freeman, M.J.; Morgan, D.; Gordon, M.N. Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J. Neuroimmune Pharmacol. 2007, 2, 222–231.
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200.
- McGeer, E.G.; McGeer, P.L. Innate immunity in Alzheimer’s disease: A model for local inflammatory reactions. Mol. Interv. 2001, 1, 22–29.
- Rogers, J.; Lue, L.F. Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer’s disease. Neurochem. Int. 2001, 39, 333–340.
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48.
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7.
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772.
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331.
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019, 15, 158–167.
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76.
- Hunt, A.; Schönknecht, P.; Henze, M.; Seidl, U.; Haberkorn, U.; Schröder, J. Reduced cerebral glucose metabolism in patients at risk for Alzheimer’s disease. Psychiatry Res. 2007, 155, 147–154.
- Mosconi, L.; Tsui, W.H.; Herholz, K.; Pupi, A.; Drzezga, A.; Lucignani, G.; Reiman, E.M.; Holthoff, V.; Kalbe, E.; Sorbi, S.; et al. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. J. Nucl. Med. 2008, 49, 390–398.
- Kapasi, A.; Yu, L.; Petyuk, V.; Arfanakis, K.; Bennett, D.A.; Schneider, J.A. Association of small vessel disease with tau pathology. Acta Neuropathol. 2022, 143, 349–362.
- Wang, M.; Iliff, J.J.; Liao, Y.; Chen, M.J.; Shinseki, M.S.; Venkataraman, A.; Cheung, J.; Wang, W.; Nedergaard, M. Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. J. Neurosci. 2012, 32, 17948–17960.
- Wang, M.; Ding, F.; Deng, S.; Guo, X.; Wang, W.; Iliff, J.J.; Nedergaard, M. Focal Solute Trapping and Global Glymphatic Pathway Impairment in a Murine Model of Multiple Microinfarcts. J. Neurosci. 2017, 37, 2870–2877.
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease–lessons from pathology. BMC Med. 2014, 12, 206.
- Laing, K.K.; Simoes, S.; Baena-Caldas, G.P.; Lao, P.J.; Kothiya, M.; Igwe, K.C.; Chesebro, A.G.; Houck, A.L.; Pedraza, L.; Hernández, A.I.; et al. Cerebrovascular disease promotes tau pathology in Alzheimer’s disease Alzheimer’s Disease Neuroimaging Initiative. Brain Commun. 2020, 2, fcaa132.
- Esiri, M.M.; Nagy, Z.; Smith, M.Z.; Barnetson, L.; Smith, A.D. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet 1999, 354, 919–920.
- Arvanitakis, Z.; Capuano, A.W.; Leurgans, S.E.; Bennett, D.A.; Schneider, J.A. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: A cross-sectional study. Lancet Neurol. 2016, 15, 934–943.
- Arvanitakis, Z.; Capuano, A.W.; Leurgans, S.E.; Buchman, A.S.; Bennett, D.A.; Schneider, J.A. The Relationship of Cerebral Vessel Pathology to Brain Microinfarcts. Brain Pathol. 2017, 27, 77–85.
- Kapasi, A.; Schneider, J.A. Vascular contributions to cognitive impairment, clinical Alzheimer’s disease, and dementia in older persons. Biochim. Biophys. Acta 2016, 1862, 878–886.
- Brown, R.; Benveniste, H.; Black, S.E.; Charpak, S.; Dichgans, M.; Joutel, A.; Nedergaard, M.; Smith, K.J.; Zlokovic, B.V.; Wardlaw, J.M. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc. Res. 2018, 114, 1462–1473.
- Boyle, P.A.; Yu, L.; Leurgans, S.E.; Wilson, R.S.; Brookmeyer, R.; Schneider, J.A.; Bennett, D.A. Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Ann. Neurol. 2019, 85, 114–124.
- Maclullich, A.M.; Wardlaw, J.M.; Ferguson, K.J.; Starr, J.M.; Seckl, J.R.; Deary, I.J. Enlarged perivascular spaces are associated with cognitive function in healthy elderly men. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1519–1523.
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385.
- Michaud, J.P.; Bellavance, M.A.; Préfontaine, P.; Rivest, S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 2013, 5, 646–653.
- Droujinine, I.A.; Perrimon, N. Defining the interorgan communication network: Systemic coordination of organismal cellular processes under homeostasis and localized stress. Front. Cell Infect. Microbiol. 2013, 3, 82.
- Palmqvist, S.; Tideman, P.; Cullen, N.; Zetterberg, H.; Blennow, K.; Alzheimer’s Disease Neuroimaging Initiative; Dage, J.L.; Stomrud, E.; Janelidze, S.; Mattsson-Carlgren, N.; et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat. Med. 2021, 27, 1034–1042.
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111.
- Julius, S.; Gudbrandsson, T.; Jamerson, K.; Andersson, O. The interconnection between sympathetics, microcirculation, and insulin resistance in hypertension. Blood Press. 1992, 1, 9–19.
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Investig. 2017, 127, 3210–3219.
- Rahimi, J.; Kovacs, G.G. Prevalence of mixed pathologies in the aging brain. Alzheimers Res. Ther. 2014, 6, 82.
- Mestre, H.; Kostrikov, S.; Mehta, R.I.; Nedergaard, M. Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin. Sci. 2017, 131, 2257–2274.
- Ishida, K.; Yamada, K.; Nishiyama, R.; Hashimoto, T.; Nishida, I.; Abe, Y.; Yasui, M.; Iwatsubo, T. Glymphatic system clears extracellular tau and protects from tau aggregation and neurodegeneration. J. Exp. Med. 2022, 219, e20211275.
- De Reuck, J.; Deramecourt, V.; Cordonnier, C.; Pasquier, F.; Leys, D.; Maurage, C.A.; Bordet, R. The incidence of post-mortem neurodegenerative and cerebrovascular pathology in mixed dementia. J. Neurol. Sci. 2016, 366, 164–166.
- James, B.D.; Wilson, R.S.; Boyle, P.A.; Trojanowski, J.Q.; Bennett, D.A.; Schneider, J.A. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain 2016, 139, 2983–2993.
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527.
- Beach, T.G.; Malek-Ahmadi, M. Alzheimer’s Disease Neuropathological Comorbidities are Common in the Younger-Old. J. Alzheimers Dis. 2021, 79, 389–400.
- Uchikado, H.; Lin, W.L.; DeLucia, M.W.; Dickson, D.W. Alzheimer disease with amygdala Lewy bodies: A distinct form of alpha-synucleinopathy. J. Neuropathol. Exp. Neurol. 2006, 65, 685–697.
- Nedergaard, M.; Goldman, S.A. Glymphatic failure as a final common pathway to dementia. Science 2020, 370, 50–56.
- Banks, W.A. The blood-brain barrier as an endocrine tissue. Nat. Rev. Endocrinol. 2019, 15, 444–455.
- Castellani, G.; Schwartz, M. Immunological Features of Non-neuronal Brain Cells: Implications for Alzheimer’s Disease Immunotherapy. Trends Immunol. 2020, 41, 794–804.
- Kim, H.; Li, Q.; Hempstead, B.L.; Madri, J.A. Paracrine and autocrine functions of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in brain-derived endothelial cells. J. Biol. Chem. 2004, 279, 33538–33546.
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483.
- Coleman, B.M.; Hill, A.F. Extracellular vesicles—Their role in the packaging and spread of misfolded proteins associated with neurodegenerative diseases. Semin. Cell Dev. Biol. 2015, 40, 89–96.
- Garcia-Contreras, M.; Thakor, A.S. Extracellular vesicles in Alzheimer’s disease: From pathology to therapeutic approaches. Neural Regen. Res. 2023, 18, 18–22.
- Alexandrov, P.N.; Dua, P.; Hill, J.M.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. microRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int. J. Biochem. Mol. Biol. 2012, 3, 365–373.
- Su, H.; Rustam, Y.H.; Masters, C.L.; Makalic, E.; McLean, C.A.; Hill, A.F.; Barnham, K.J.; Reid, G.E.; Vella, L.J. Characterization of brain-derived extracellular vesicle lipids in Alzheimer’s disease. J. Extracell. Vesicles 2021, 10, e12089.
- Ruan, Z.; Pathak, D.; Venkatesan Kalavai, S.; Yoshii-Kitahara, A.; Muraoka, S.; Bhatt, N.; Takamatsu-Yukawa, K.; Hu, J.; Wang, Y.; Hersh, S.; et al. Alzheimer’s disease brain-derived extracellular vesicles spread tau pathology in interneurons. Brain 2021, 144, 288–309.
- Ma, Q.; Xing, C.; Long, W.; Wang, H.Y.; Liu, Q.; Wang, R.F. Impact of microbiota on central nervous system and neurological diseases: The gut-brain axis. J. Neuroinflamm. 2019, 16, 53.