2. Classical Type 1 Diabetes Mellitus Pathoetiology
T1DM arises from the dysregulation of pancreatic β-cell insulin production and insulin effects. Pancreatic β-cell function and insulin release are intimately linked to variations in blood glucose. At low/basal levels of blood glucose, the ATP-sensitive potassium (K
ATP) channels in pancreatic β-cells remain open, leading to maintained membrane hyperpolarization coupled to Ca
2+ channel closure, thereby inhibiting insulin secretion by pancreatic β-cells [
10]. When blood glucose concentration levels rise, glucose is taken up by the glucose transporter (GLUT)1 into pancreatic β-cells where it is quickly phosphorylated by glucokinase and then converted to pyruvate. The conversion of pyruvate to acetyl-CoA by the pyruvate dehydrogenase complex (PDC) increases ATP production by the tricarboxylic acid (TCA) cycle and mitochondrial oxidative phosphorylation (OXPHOS), resulting in K
ATP channel closure, plasma membrane depolarization, and the opening of voltage-dependent Ca
2+ channels in association with insulin vesicle exocytosis. Mitochondrial function is therefore a crucial aspect of pancreatic β-cell function and plasticity of response to variations in circulating glucose [
10].
Islet amyloid polypeptide (amylin) is simultaneously secreted by pancreatic β-cells along with insulin. Amylin accumulates in diabetes, forming amyloid deposits in the pancreas that are a relevant aspect of T1DM and T2DM pathophysiology, driving pancreatic β-cell dysfunction and apoptosis, as well as contributing to islet transplantation failure [
11,
12]. Amylin fibrillation follows amylin over-production in association with insulin resistance and hyperinsulinemia, which triggers a nucleation-dependent self-assembly of amylin into intracellular or extracellular amyloid deposits [
13]. Amylin fibrillation is proposed to parallel other amyloidosis, such as amyloid-β in dementia, in driving pancreatic β-cell apoptosis and may therefore be an important treatment target in T1DM [
13], including in relation to the ‘autoimmune’ aspects of T1DM. Amylin, Ca
2+ dysregulation, reactive oxygen species (ROS), endoplasmic reticulum alterations, and metabolic dysregulation have all been proposed to drive post-translational protein modifications that underpin the autoimmune-like response in T1DM [
11].
T1DM pathophysiology is classically described as a T cell-mediated autoimmune disorder, whereby predominantly CD8
+ T cells become autoreactive, leading to the destruction of pancreatic β-cells. A failure in the capacity of regulatory T cells (Treg) to suppress such processes is also implicated. Elevated levels of major histocompatibility complex (MHC) class 1 molecules on pancreatic β-cells drive the autoreactivity responses of CD8
+ T cells. Such autoreactive CD8
+ T cells usually undergo negative selection in the thymus, which seems to be altered/impaired in T1DM due, at least partly, to an allelic variant of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) modulator, NF-κB-inhibitor Delta (Nfkbid), which is an atypical NF-κB inhibitor and an important regulator of B cell immunity across a host of medical conditions [
14]. Modulation of Nfkbid thymic expression levels indicates that it may act to regulate levels of both autoreactive CD8
+ T cells and Treg [
15,
16].
T1DM susceptibility is linked to several genetic and environmental factors. Familial transmission is apparent in about 10% of T1DM patients, with the human leukocyte antigen complex (HLA) located on chromosome 6 showing the strongest familial association [
17]. Numerous single nucleotide polymorphisms (SNPs) have been linked to T1DM risk, including P53 [
18], the NF-κB modifying gene, small ubiquitin-related modifier 4 (SUMO4) [
19], and many other genes [
20] as well as HLA haplotypes [
21]. Many T1DM susceptibility genes act to regulate mitochondrial function and mitophagy in pancreatic β-cells, including Clec16a [
22], highlighting the importance of optimal mitochondrial function and OXPHOS [
23] in the regulation of pancreatic β-cell function and insulin regulation.
T1DM environmental risk factors include the consumption of a Western-type diet, with a number of ligands for the receptor for advanced glycation end-products (RAGE) contained within many such foods [
24]. These authors propose that RAGE activation leads to the activation and proliferation of islet infiltrating CD8
+ and CD4
+ T cells, coupled to Treg suppression, thereby impacting on the patterned immune response that contributes to pancreatic β-cell injury, with effects mediated via NF-κB upregulation, coupled to increased ROS and oxidative stress [
24]. RAGE has numerous other ligands, including high-mobility group box (HMGB)1 and S100/calgranulin family members, such as S100A9, as well as amyloid-β and pre-fibrillar amylin aggregates [
25,
26]. Preclinical data show that the administration of soluble RAGE dramatically suppresses T1DM via Treg upregulation, and associated inhibition of conventional T cell division [
27]. Such data would implicate a significant clinical impact of RAGE ligands that can be suppressed by soluble RAGE, as well as melatonin [
28]. The antioxidant, anti-inflammatory, circadian, and mitochondrial optimizing effects of melatonin are important aspects of T1DM, whilst the immediate precursor of melatonin,
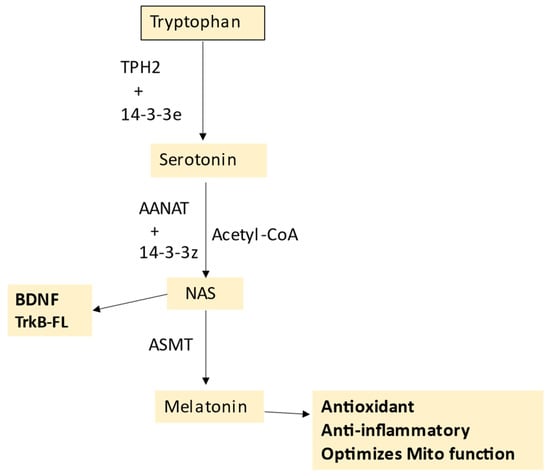
N-acetylserotonin (NAS) may also be a crucial regulator of pancreatic β-cell survival, via its capacity to activate the brain-derived neurotrophic factor (BDNF) receptor TrkB, as shown in
Figure 1. The role of the melatonergic pathway in T1DM is detailed in
Section 5.
Figure 1. The tryptophan–melatonin pathway. Tryptophan is converted to serotonin by tryptophan hydroxylase (TPH)1 or TPH2, which requires stabilization by 14-3-3e. AANAT converts serotonin to N-acetylserotonin (NAS), with AANAT requiring stabilization by another 14-3-3 isoform and the presence of acetyl-CoA as a co-substrate. ASMT converts NAS to melatonin. NAS is a BDNF mimic, via its activation of the BDNF receptor, TrkB. NAS may also induce BDNF. Melatonin is a powerful antioxidant and anti-inflammatory that optimizes mitochondrial function. Abbreviations: AANAT: aralkylamine N-acetyltransferase; Acetyl-CoA: acetyl coenzyme A; ASMT: N-acetylserotonin O-methyltransferase; BDNF: brain-derived neurotrophic factor; NAS: N-acetylserotonin; TPH: tryptophan hydroxylase.
However, many of the genetic and epigenetic susceptibility factors for T1DM may ultimately act on mitochondrial function in pancreatic β-cells [
23], immune cells, and other cells across the body.
3. Wider T1DM Pathophysiology
A wide array of diverse signaling pathways, processes, and factors are associated with T1DM pathoetiology and pathophysiology.
The inability to prevent pancreatic β-cell loss and to induce regeneration, coupled with the complexity of immune-mediated processes underpinning autoimmune-like changes, has generated the investigation of a wide range of possible pathophysiological processes and factors in T1DM, including the aryl hydrocarbon receptor (AhR) [
29], toll-like receptor (TLR)4 activation [
30], NF-κB [
31], yin yang (YY)1 [
32], the melatonergic pathway [
33], P53 [
18], circadian dysregulation [
34], and gut dysbiosis/permeability [
35].
The AhR is predominantly expressed in a complex with other proteins in the cytoplasm, with its activation by endogenous and exogenous ligands leading to its translocation to the nucleus, where it forms a dimer that upregulates genes containing the xenobiotic response element. The AhR has complex effects that are partly dependent upon the specific AhR-activating ligand, cell type, and concurrent wider cellular processes [
36]. The AhR is also expressed on the mitochondrial membrane where it can regulate Ca
2+ influx via the voltage-dependent anion channel (VDAC)1 [
37]. The AhR has diverse effects in different cells and tissues relevant to T1DM pathophysiology, with AhR activation leading to suppressed function and cytotoxic capacity in CD8
+ T cells and natural killer (NK) cells [
38], the maintenance of the gut barrier [
39], and pancreatic β-cell regulation [
40]. The diverse, and sometimes contrasting, effects of AhR activation in different cells and tissues by various ligands complicate its incorporation into T1DM pathophysiology.
Western diet-driven increases in palmitate and lipopolysaccharide (LPS) can synergistically damage pancreatic β-cells via TLR4 activation, including via enhanced pancreatic β-cell ceramide and suppressed sphingosine-1-phosphate (S1P) levels [
30]. The role of TLR4 signaling in pancreatic β-cell dysfunction has been widely replicated [
41], with factors acting to regulate TLR signaling, including miR-383 and TLR4 knockout, ameliorating high fat/sugar diet-induced β-cell dysfunction, as mostly assessed in T2DM [
42,
43]. The protection afforded by melatonin in pancreatic β-cells is at least partly mediated via TLR2/4 level suppression [
44]. Such data also indicate the relevance of increased gut permeability to pancreatic β-cell dysfunction via raised circulating LPS levels [
45], with the association of palmitate with pancreatic β-cell dysfunction partly mediated via increased gut permeability [
46].
LPS activation of TLR4 induces the transcription factors, NF-κB and YY1, in many cell types, including leukocytes, macrophages, microglia, and astrocytes [
36,
47]. The induction of pro-inflammatory processes in pancreatic β-cells is intimately linked to NF-κB upregulation [
31,
48,
49]. The downstream consequences of NF-κB upregulation are associated with a diverse array of factors, including non-steroidal anti-inflammatory drug activated gene-1, growth differentiation factor-15 (NAG-1/GDF15) [
48], or intercellular adhesion molecule (ICAM)-1 [
31]. YY1 knockout in pancreatic β-cells leads to rapid hyperglycemia onset, impaired glucose tolerance, and suppressed pancreatic β-cell mass, both in neonates and adult murine models [
32]. These authors showed YY1 to bind the enhancer regions in exon 2 of Ins1 and Ins2, activating pro-insulin and insulin transcription and thereby insulin production from pancreatic β-cells [
32].
Such data on NF-κB and YY1 indicate contrasting effects in T1DM pathophysiology. However, it is important to note that both transcription factors can induce the melatonergic pathway, as shown in different cell types [
50,
51]. Variation in the capacity of these transcription factors to upregulate the melatonergic pathway is of some importance to T1DM pathophysiology, given that melatonin protects and optimizes pancreatic β-cell function and insulin regulation at nM levels [
33,
52], including via the optimization of mitochondrial function. This may be of relevance to data showing that the induction and activation of NF-κB may afford protection against pancreatic β-cell loss in T1DM models, which the authors attribute to NF-κB induction of miR-150, thereby preventing T1DM-associated inflammation and pancreatic β-cell apoptosis [
53]. As NF-κB is associated with the induction of the melatonergic pathway in several cells so far investigated [
51,
54], it is of note that the most commonly used preclinical T1DM model, the streptozotocin-induced T1DM model, suppresses local melatonin production and melatonergic pathway activity, as shown in the retina [
55]. This could indicate that the suppression of the mitochondrial melatonergic pathway may be a significant factor in pancreatic β-cell apoptosis in clinical T1DM, which is elaborated upon in
Section 7.
Other intracellular signaling processes and transcription factors are associated with T1DM, including SNPs in, and upregulation of, the classical tumor suppressor and transcription factor, P53 [
18]. However, although P53 is upregulated during apoptosis in pancreatic β-cell in the course of T1DM and T2DM, recent data indicate that P53 is not essential to pancreatic β-cell loss [
56]. Using pancreatic β-cell specific P53 knockout, these authors showed pancreatic β-cell specific P53 knockout failed to ameliorate insulin secretion and glucose tolerance, as well as failing to raise pancreatic β-cell numbers in an array of genetic, dietary, and pharmacological models of T1DM and T2DM [
56]. These authors suggest that the induction of poly [ADP-ribose] polymerase 1 (PARP-1) may be a more relevant mediator of pancreatic β-cell loss in T1DM models [
56], by decreasing NAD
+ availability, thereby suppressing the sirtuin-induced PDC and mitochondrial OXPHOS.
Circadian dysregulation, both genetic and environmental, is closely associated with metabolic syndrome and T2DM, involving the uncoupling of OXPHOS, ATP production, and glucose-stimulated insulin secretion [
34], mediated—in part—via the suppression of the circadian gene, Bmal1, and the endogenous antioxidant inducing transcription factor, Nrf2 [
34]. These changes can be reversed by melatonin [
57,
58], highlighting the importance of pineal melatonin in the circadian optimization of pancreatic β-cell function. Circadian dysregulation is also intimately linked to a diverse array of T1DM symptomatology [
59], including nocturnal non-dipping blood pressure increasing kidney disease [
60], cardiac autonomic neuropathy [
61], platelet morphology [
62], microvascular complications [
63], and patterned immune activity [
64], whilst circadian variation in basal insulin requirement can be an early marker of autoimmune polyendocrine syndromes in T1DM [
65]. Such data highlight the role of alterations in the circadian rhythm in T1DM pathophysiology.
The growing appreciation of the role of the gut microbiome and gut permeability across a host of diverse medical conditions is also highly relevant in T1DM [
66,
67]. The role of microbiota in T1DM and T2DM is highlighted by data showing the impact of the maternal endometrial and/or vaginal microbiomes of diabetic mothers, including in gestational diabetes mellitus, which can act through epigenetic mechanisms to increase T2DM—and possibly T1DM—risk in the offspring [
67]. Such data highlight the importance of microbiota in the regulation of metabolism and the likelihood of prenatal priming of later T1DM related processes in pancreatic β-cells. This requires future investigation.
Recent data causal models the beneficial effects of human umbilical cord mesenchymal stem cell vesicles containing exenatide as being mediated not only directly in pancreatic β-cells but also via alterations in the gut microbiome/permeability [
68]. The beneficial effects of
Lactobacillus johnsonii strain N.6 nanovesicles in T1DM are proposed to be mediated via the upregulation of AhR ligands and AhR activation, with effects in both pancreatic β-cells as well as in macrophages, which are induced into a M2b-like phenotype [
69]. A number of preclinical studies over the past decade have shown the beneficial effects of
Lactobacillus johnsonii in delaying T1DM onset, which is proposed to be mediated by a number of processes, including suppression of Th17 cells, increasing intestinal crypt Paneth cell numbers, and suppressing gastro-intestinal caspase-1 induction [
70,
71,
72], whilst also decreasing the kynurenine/tryptophan ratio, increasing cytotoxic CD8
+ T cells, and changing the patterned immune response, as shown in healthy human volunteers [
73]. Whether
Lactobacillus johnsonii, and other bacteria, in the gut drive the induction of AhR ligands (such as indole-3-propionate) will be important to determine.
Lactobacillus johnsonii also increases the short-chain fatty acids, butyrate, propionate, and acetate, indicating wider effects of gut microbiome-derived products, including butyrate’s epigenetic effects as a histone deacetylase (HDAC) inhibitor [
74]. HDACi, via PKA upregulation and tryptophan hydroxylase (TPH)1 induction, derepresses serotonin production, thereby potentiating pancreatic β-cell function [
75]. As butyrate optimizes mitochondrial function with effects that involve the upregulation of the melatonergic pathway, as shown in intestinal epithelial cells [
76], alterations in the capacity to upregulate the melatonergic pathway in pancreatic β-cells will be important to determine. It clearly requires investigation as to the relevance of gut microbiome-derived butyrate, including as regulated by
Lactobacillus johnsonii, in the pathoetiology of T1DM and the importance of a functional tryptophan–melatonin pathway in pancreatic β-cells. The full effects of butyrate require the capacity of a cell to upregulate the mitochondrial melatonergic pathway [
76].
Importantly, the gut microbiome is comprised of not only bacteria, but also fungi and viruses, including enteroviruses and bacteriophages, with all these groupings showing changes at the initiation of pediatric T1DM [
77,
78]. Investigations on the gut microbiome in T1DM, versus controls, have focused on changes in gut bacteria, showing elevations in
Prevotella copri and
Eubacterium siraeum, with relative attenuation of
Firmicutes bacterium and
Faecalibacterium prausnitzii [
79] in T1DM patients. Other studies have investigated a wider range of changes in the gut microbiome, indicating no significant differences in α-diversity between T1DM and controls [
80]. However, these authors found T1DM patients to have 43 bacterial taxa significantly depleted and 37 bacterial taxa significantly enriched [
80]. This study also found disease duration and glycated hemoglobin (HbA1c) to explain a significant part of the gut microbiome variation in T1DM, whilst neuropathy and macrovascular complications were significantly linked to variations in several microbial species [
80]. However, as noted by the authors of these studies [
79,
80], and other studies [
81], the mechanistic links to pancreatic β-cell loss and wider T1DM pathophysiology remain to be determined.
Limited data on bacteriophages in the pathoetiology of T1DM indicate that amyloid-producing
Escherichia coli (
E. coli),
E. coli phages, and bacteria-derived amyloid may be involved in the early stages of T1DM pathoetiology, as indicated in data derived from children at high risk of T1DM [
82]. This study and other data indicate that changes in the gut virome may precede the initial signs of T1DM, and therefore be of relevance to T1DM pathoetiology [
83]. The causative relationship has still to be determined. However, such data may indicate that the gut virome may be more relevant to T1DM pathoetiology than gut bacteria, which tend to show diversity only after the emergence of T1DM. Enteroviruses are one of the major environmental triggers of childhood-onset T1DM, with recent data indicating that enteroviruses may also be an important trigger in adult-onset T1DM [
84]. Overall, data indicate an interrelatedness of the gut microbiota, metaproteome, and virome that is relevant to T1DM onset, as investigated in young children, with a functional remodeling of the gut microbiota accompanying islet autoimmunity [
77]. An initial bacteriophage or enterovirus impact on gut microbiome diversity seems followed by a decrease in butyrate-producing bacteria, with consequences for mitochondrial function systemically. As to whether the suppressed butyrate and/or other gut bacteria products are drivers of changes in pancreatic β-cells, either directly or indirectly suppressing the mitochondrial melatonergic pathway in pancreatic β-cells requires investigation.
Interestingly,
Candida albicans fungi is elevated in T1DM, including at the time of initial presentation [
85]. As
Lactobacillus johnsonii can eliminate
Candida albicans fungi from the gut [
86], the clinical benefits of
Lactobacillus johnsonii are likely to include alterations in the wider gut microbiome. However, it still requires clarification as to whether the gut is a primary site of change or whether the emergence of fungal infections is driven by a suppressed anti-fungal immune response [
87], which may also involve alterations in the gut microbiome [
67]. Two fatty acids produced by
Lactobacillus johnsonii (and
Bacteroides thetaiotaomicron), namely oleic acid and palmeic acid, mediate many of
Lactobacillus johnsonii benefits, including within the gut and immune cells, indicating that gut microbiome-associated regulation of the immune response is a relevant aspect of T1DM pathoetiology [
88]. Future research will have to clarify the relevance of
Lactobacillus johnsonii specific effects in T1DM, including in interaction with
Candida albicans fungal infection.