Along the years, radiotherapy (RT) has largely contributed to better disease control, reducing nontargeted tissue toxicity and improving overall survival through the use of conformal and intensity-modulated RT or charged particles. However, the appearance of local recurrences and metastases after RT has underlined the need for additional therapies. The last decade saw the progressive approval of monoclonal antibodies targeting negative costimulatory immune receptors known as immune-checkpoint inhibitors (ICIs), which achieved considerable therapeutic success in a certain number of advanced cancers. A better understanding of the effects of RT dose and fractionation allowed uncovering the immunological effects of RT and developing combined therapies between RT and immunotherapy. With the approval of the first PARP inhibitor (PARPi) in 2014, new therapeutic tools targeting DNA repair mechanisms have progressively become available, and synergies with RT and immunotherapy have been explored.
Radiotherapy plays a major role in the treatment of a wide range of malignancies. Around 60–70% of patients undergo treatments, mostly with photon therapy (X- or γ-rays), in addition to others with heavy ions and protons.
Ionizing radiations (IRs) induce DNA damage with the number, distribution, variety, and severity of lesions depending on the quality of radiation, dose, fractionation, cell physiological status, and tumor microenvironment (TME) (including oxygen availability). DNA damage is induced by direct energy deposition or indirectly through the generation of highly reactive free radicals. Lesions induced by a single ionizing trajectory localize within short distances (nanometers); these clustered lesions are a typical signature of IR-damaged DNA [
3,
4]. The large number of clustered DNA lesions, including multiple double-strand breaks, generated by IR are hardly fixed by the DNA repair mechanisms and lead to cell-cycle arrest and/or cell death [
5].
While radiation toxicity has been considered the (main) mechanism of action in radiotherapy, its collateral tissue damage has urged more favorable ratios between the dose adsorbed by the targeted tumor and the normal tissues. Both three-dimensional conformal and intensity-modulated RT reduced nontargeted tissue toxicity and improved overall survival compared with two-dimensional RT, although with some contrasting conclusions [
6,
7]. A better dose distribution on the targeted tissue compared with photon RT can be obtained with charged particles. While traveling through tissues, protons and carbon ions, the most used charged particles for RT, release energy according to a typical curve ending with a pronounced peak (Bragg peak). At the Bragg peak, the majority of the energy is released (with a tiny lateral scatter) and a massive ionization of the surrounding matter occurs. Tissues lying beyond the Bragg peak are, therefore, spared. Superimposition of multiple Bragg peaks spanning the tumor volume improves disease control [
8].
2.1. Immuno-Stimulating Effects of Radiotherapy
During the last two decades, it has become progressively evident that IRs do not exert their effects exclusively by direct killing of tumor cells. Radiotherapy has important immunological effects by inducing the expression of IFNs, as well as other cytokines and chemokines, the release of tumor-associated antigens (TAA), immunogenic cell death (ICD), and changes in the TME [
11,
12].
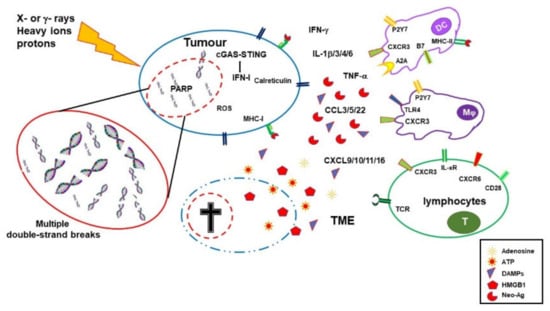
IR-induced leakage of nuclear and mitochondrial DNA into the cytosol activates the cyclic GMP–AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway. cGAS/STING signaling, a pathway normally involved in antiviral responses, results in the expression of type I IFN (IFN-I) in irradiated cells and sustains the antitumor immune response [
13,
14]. IFN-I, together with other signals, promotes recruitment and activation of dendritic cells (DCs), which in turn activates CD8 cells to perform T-cell killing, a process essential for tumor reduction [
15]. Noteworthily, experiments comparing equivalent doses of photon, proton, and carbon ion IRs showed that, despite differences at early timepoints, all these radiotherapeutic agents induced a similar gene expression signature in exposed tumor cells involving the activation of the GAS/STING pathway and STAT1-dependent responses [
16].
In addition to the upregulation of IFN-I, RT induces the expression of several cytokines and chemokines, consequently orchestrating recruitment and activation (or suppression, see below) of several leukocyte populations into the tumor site. RT-induced cytokines include IFNγ, IL1β, TNFα, IL-3, IL-4, IL-6, and TGFβ [
17]. Cytokines and chemokines are known to mutually regulate their expression. RT-induced IL1β expression, for instance, upregulates CCL2 production and, consequently, sustains the recruitment of CCR6
+ monocytes and T cells [
18]. Upregulation of several chemokines, including CCL3, CCL5, CCL22, CXCL9, CXCL10, and CXCL11, has been described to play different roles with effects depending on tumor type and other TME factors [
19,
20]. CXCL16 is also upregulated by IRs in both mouse and human breast cancer cells, representing a major factor in driving CXCR6-expressing Th1 and CD8 T cells to the tumor site [
21].
RT-injured tumor and tumor-infiltrating cells release intracellular molecules known as damage-associated molecular patterns (DAMPs), or alarmins, including high-mobility group box 1 (HMGB1), ATP, and calreticulin. DAMPs are released through both passive mechanisms, due to cell damage-associated leakage, and different active processes, depending on the stressing stimulus, which includes RT-induced reactive oxygen species (ROS) generation. Through specific receptors, DAMPs are recognized as danger signals by immune and nonimmune cells, resulting in inflammatory response, with the release of chemotactic factors, upregulation of adhesion molecules, and leukocyte recruitment and activation. Danger signals, therefore, generate the immunogenic context promoting immune responses toward TAA released by RT-damaged cells in the TME [
24].
HMGB1, passively released by dying cells or actively secreted by inflammation-stimulated cells, is recognized by Toll-like receptor 4 (TLR4) and by the receptor for advanced glycation end-products (RAGE), both expressed on several cells, including macrophages and dendritic cells. TLR4 and RAGE engagement by HMGB1 leads to NF-κB activation and expression of proinflammatory cytokines, release of chemotactic factors, and recruitment and activation of leukocytes [
25].
The concentration of ATP in the extracellular space modulates different functions including cell differentiation, proliferation, adhesion, and death. Any type of cell death induces secretion or release of ATP, although the involved mechanisms depend on the type of death stimulus and the apoptotic stage. Extracellular ATP is perceived as a “find me” signal which drives macrophages to the dying cells through P2Y2 receptors [
24]. However, when present at higher concentrations, the ATP can also be recognized by the purinergic P2X7 receptors on dendritic cells and activate the NALP3-inflammasome pathway, thus acting as a danger signal and inducing immunogenic responses. ATP-stimulated dendritic cells produce IL-1 and IL-18, which synergize with IFNγ in the induction of tumor-specific CD8 T cells.
Calreticulin is a molecule mostly localized in the endoplasmic reticulum, playing several immune roles including assembly of MHC I molecules and loading of peptides on the MHC I groove. Calreticulin is involved in cell signaling, Ca
2+ homeostasis, and cell migration and proliferation [
28]. Production of ROS and reactive nitrogen species (RNS), induced by photon or proton radiotherapy, leads to endoplasmic reticulum stress and calreticulin exposure on the external cell membrane [
29,
30].
Photon radiotherapy increases the expression of MHC class I molecules on tumor cells, a finding that was also observed, more recently, using protons [
30]. RT induces the expression of novel proteins and neoantigens, as well as enhances protein degradation and the generation of additional peptides, which are presented to CD8 T cells in association with MHC class I molecules. These two properties may cooperate to increase antigen presentation and activation of tumor specific CD8 immune responses.
Radiotherapy, therefore, by inducing the release of DAMPs, the expression of cytokines and chemokines, cell death, and the release/expression of TAA, has the potential to create an inflammatory/immunogenic context where innate and TAA-specific adaptive immune cells could be activated and generate an antitumor immune response (
Figure 1), thus providing the rationale for radioimmunological synergic therapies [
37,
38].
Figure 1. Immuno-stimulating effects of radiotherapy. Ionizing radiations (IRs) induce DNA damage, oxidative stress, and cell death. Dying cells release HMGB1 and ATP, and then expose calreticulin on their cell surface, all these molecules being features of immunogenic cell death. Damaged DNA activates the cGAS/STING pathway, leading to IFN-I expression, upregulation of MHC I expression, and improved antigen presentation in surviving cells. IRs also alter the tumor microenvironment (TME) by inducing the expression of several cytokines and chemokines with consequent recruitment of leukocytes. Tumor cells might accumulate mutations and express neoantigens which are taken up by dendritic cells (DCs) that, in the presence of the inflammatory stimuli, mature and migrate to the draining lymph node where they prime tumor antigen-specific CD4 and CD8 T cells.
2.2. Immuno-Depressing Effects of Radiotherapy
RT has been known for a long time to induce immunosuppressive effects through a toxic action. Leukopenia is one of the most frequent effects of RT. When the area of exposure includes bone marrow cells, a long time is required to fully recover the hematopoietic damage both in clinic and in experimental settings [
39,
40,
41]. However, leukocytes display different grades of susceptibility to the effects of IRs, depending on cell type, activation status, and cell-cycle phase. Myeloid cells, including monocytes/macrophages, dendritic cells, and myeloid-derived suppressor cells (MDSCs), are more resistant to IR than lymphocytes and NK cells, probably for their reduced proliferative rate compared with lymphoid cells [
42]. Within the myeloid lineage, circulating monocytes are more susceptible than tissue macrophages [
43]. Moreover, M2-polarized macrophages, which play an unfavorable role in tumor immunity, have been shown to be more resistant to IRs than M1-polarized macrophages, both in vivo and in vitro and under both normoxia and hypoxia [
44,
45].
Lymphocytes are more susceptible than myeloid cells, and they undergo apoptosis during the interphase if irradiated. B cells have been shown to be particularly susceptible to radiations [
42]. T cells are quite heterogeneous in their susceptibility, with activated T cells being more resistant than resting cells and CD4 being more resistant than CD8 T cells. Tumor-associated and tissue-resident memory T cells were described to be more resistant than naïve T cells, probably for their pre-activated status and for the protective effects of the TGFβ often present in the TME [
46]. Within the CD4 T-cell population, Foxp3-expressing regulatory T cells appear to be more resistant. Upon radiation, the percentage of Treg cells is increased within the tumor site compared with CD8 and non-Treg CD4 cells [
47,
48].
The Immune suppressive effects of IRs are also induced by prolonged and/or intense activation of signals normally associated with immune activation. Expression of inhibitory cytokines, recruitment of T and myeloid regulatory cells, and an immune-suppressive TME are associated with repeated irradiation [
52]. Irradiation-induced chronic expression of IFN-I leads to the upregulation of PD-L1, as shown in tumor cell lines and in tumor-infiltrating macrophages [
53,
54]. STING-sustained IFN-I production also results in increased expression of CCL2 and recruitment of monocytic MDSC and Treg cells [
55,
56].
3. PARP Inhibitors
PARP-1, the most abundant member of the poly(ADP-ribose) (PAR) polymerase (PARP) family, more recently defined as diphtheria toxin-like ADP-ribosyltransferases (ARTDs), accounts for the majority of PARylation activity and has a high DNA damage-sensing ability [69]. Free DNA ends activate PARP-1, which highly PARylates itself and detaches from chromatin. Indeed, addition of PARs radically changes the electric charge of the targeted molecule, rendering it highly negative. As a consequence, PARylated proteins are electrostatically repulsed by the DNA, a mechanism involved in chromatin accessibility to DNA repair enzymes (and to DNA transcription and replication regulators). PARP-1 also generates large amounts of PARs that work as scaffolds recruiting DNA repair enzymes to the lesion site, including XRCC1 [70]. PARP-1 plays a central role in orchestrating responses to genotoxic stress and represents a critical enzyme in single-strand break and alternative end-joining repair [71,72]. However, recent studies also indicated that PARP-1 plays a role in double-strand break (DSB) repair mechanisms, including homologous recombination and classical nonhomologous end-joining (c-NHEJ) [73,74].
Following a long period of preclinical and clinical studies, PARP inhibitors (PARPis) reached wide clinical use with the approval of olaparib (AZD-2281) in 2014 and later on of niraparib (MK-4827), rucaparib (AG-014699), talazoparib (BMN673), and veliparib (ABT888) for treatment of ovarian, breast, prostate, and pancreatic cancer [75,76]. PARPis are the first clinically approved drugs exploiting synthetic lethality; that is, they target a function specifically vital in mutation-bearing cancer cells [77,78]. PARPis were shown to be lethal in homologous recombination (HR)-deficient BRCA1/BRCA2-mutated cancers, likely because collapsed replication forks are no longer repaired [79,80]. However, recent preclinical and early clinical studies also sustained the use of PARPis in other molecular subsets of cancer, including cancers with high replication stress [81].
All clinically approved PARPis share a nicotinamide-based moiety that inhibits PARP-1 enzymatic activity by competing for binding to the catalytic site with NAD. PARPis prevent PARP-1 auto-PARylation and its consequent removal from chromatin and DNA lesions. This effect, termed PARP trapping, is currently the preferred interpretative model of the PARPis mechanism of action. Indeed, cytotoxicity due to PARP correlates with the ability to trap PARP on DNA lesions and is more cytotoxic than gene deletion. PARP trapping leads to replication fork collapse during the S phase and consequent cell death [82,83].
4. Synergy between PARPi and Radiotherapy
Although PARPis represent an unprecedented success in cancer chemotherapy, the therapeutic response ranges between 30% and 50%, and tumors develop resistance during treatment, urging additional solutions. On the other hand, tumors can also become resistant to radiotherapy, often through alterations in DNA repair pathways, with this possibility being reduced by combined chemotherapy. Noteworthily, several radioresistant tumors express PARP-1 at high levels [
90,
91]. In tumors exposed to IRs, PARPis could compromise DNA repair, hampering both SSB and DSB resolution and leading to DNA replication fork collapse. Although the radio-sensitizing effect is expected to be higher in BRCA1/BRCA2-mutated cancers, PARPis were shown to synergize with RT regardless of the HR proficiency [
92,
93]. As PARPis exert their synergic effects with RT during the S phase of the cell cycle, they could render tumor cells more susceptible to RT than nontumor slowly/nonproliferating tissue cells [
94].
Hypoxia in the tumor microenvironment activates mechanisms of adaptation in tumor cells through the hypoxia-inducible factors (HIFs), which transcriptionally activate genes guiding cellular metabolism, angiogenesis, metastasis, and other processes. As IRs induce large DNA damage through the generation of ROS, hypoxia limits their effects and results in resistance to radiotherapy. In response to hypoxia, PARP-1 regulates the stability and the activity of both HIF1 and HIF2, promoting tumor cell survival. Consistently, inhibition of PARP has been shown to control tumor growth by dampening HIF activation [
95,
96]. Thus, PARPis could also exert a synergic therapeutic effect with RT through this mechanism.
5. Synergic Immunological Effects of RT and PARPi
Beyond DNA repair, several studies have shown that PARP-1 plays a relevant role in inflammation and immune responses by regulating the activation and differentiation of both innate and adaptive immune cells. Indeed, PARPis induce several immunological effects, some of which can be detrimental in cancer therapy, while others are beneficial [
99,
100].
Impairment of DNA repair, due to either mutations or PARPi therapy, can further sustain the damaged DNA-induced activation of the cGAS/STING pathway. Clinically approved PARPis have been shown to induce IFN-I and CCL5 expression in tumor cells trough cGAS-STING [
101]. As it occurs with RT [
102], the activation of this pathway by PARPis leads to CD8 T-cell recruitment at the tumor site, with the effect being more pronounced in HR-deficient triple-negative breast cancer [
103]. Increased IFNγ and TNFα production by CD8 T cells and NK cells was also observed in a BRCA1-deficient ovarian cancer model upon treatment with PARPis [
104]. In this model, a reduction in the frequency of MDSCs, which negatively regulate antitumor immune responses, was also induced by PARP inhibition [
104].
As discussed above, the described effects on cytokine production, cell recruitment, and mutational burden could be induced by both PARPis and RT, with synergistic effects being more likely to occur in DNA damage response-deficient tumors. In an EGFR-mutated NSCLC mouse model, niraparib increased the RT driven antitumor immunity by upregulating IFN-I production through a synergic effect on the cGAS/STING pathway. The observed reduced tumor growth and prolonged survival was associated with increased CD8 T-cell infiltration [
111].
Combined PARPis and RT, therefore, have the potential to induce inflammatory signals and immunogenic cell death, as well as activate innate immune cells, consequently creating the context for the activation of the adaptive immune response toward TAA. Noteworthily, the effects are expected to be higher in genomic unstable/DNA repair compromised tumor cells, in which a wider TAA repertoire might also be generated. Effects on the expression of adhesion molecules and other alterations in the TME could contribute to immune cell recruitment and, therefore, might be useful in the treatment of tumors with a low/absent tumor immune infiltration. However, whether immune-stimulating factors induced by combined PARPis-RT prevail over suppressive elements could be sustained by synergies with further therapeutic agents.
6. Immune-Checkpoint Inhibitors
During last 30 years, seminal publications and subsequent studies by James Allison and Tasuku Honjo, both receiving the 2018 Nobel Prize in Physiology and Medicine [
118], fostered a large wealth of studies in preclinical models and clinical trials on the use of CTLA-4 (CD152) and PD-1/PD-L1 (CD279/CD274) immune-checkpoint inhibitors (ICI) in cancer therapy.
CTLA-4, an immunoglobulin gene superfamily member discovered in activated CD8 T cells more than 30 years ago [
119], is a receptor that negatively regulates cell proliferation, cytokine production, and cytotoxic functions in T cells through several mechanisms [
120,
121,
122,
123]. CTLA-4 blockade, i.e., the use of antagonist antibodies preventing CTLA-4 engagement by the natural ligands CD80 (B7.1) and CD86 (B7.2), was soon explored as a therapeutic target in tumor models by Allison [
124].
PD-1, initially discovered in activated T cells by Honjo [
128], belongs to the Ig gene superfamily and is also expressed in B and NK cells, as well as in activated macrophages and dendritic cells. Stimulation of PD-1 by either PD-L1 or PD-L2 ligands negatively regulates T-cell-mediated responses including cytokine production, cell proliferation, and cytotoxic activity, although through mechanisms different from CTLA-4 [
129,
130]. PD-L1 and PD-L2 are expressed by antigen-presenting cells and stromal cells, and they play a relevant role in maintaining immune tolerance. PD-L1 is also expressed by some tumor cells, tumor-infiltrating leukocytes, and tumor-associated fibroblasts. PD-1 engagement on tumor-infiltrating T cells by PD-L1 inhibits their cytotoxic action toward tumor cells and leads to T-cell exhaustion, favoring tumor immune evasion [
131,
132].
CTLA-4 mainly acts in the control of T-cell activation and consequent effector T-cell generation, contributing to the maintenance of immune tolerance. Antagonistic antibodies toward CTLA-4 lower the threshold for T-cell activation and sustain the expansion of antigen-stimulated T cells, a mechanism underlying their therapeutic and toxic effects. The response can indeed be to antigens expressed on tumor and normal cells [
136].
The anti-CTLA-4 antibody ipilimumab (Yervoy) was the first approved ICI recommended for the therapy of melanoma in 2011, followed a few years later by the anti-PD-1 nivolumab for non-small-cell lung cancer (NSCLC). During the following decade, several other ICIs targeting CTLA-4 (tremelimumab in October 2022), PD1 (pembrolizumab, cemiplimab, and dostarlimab), and PD-L1 (atezolizumab, avelumab, and durvalumab) were progressively approved for clinical use as single or combined therapies.
ICI achieved considerable therapeutic success in a certain number of (advanced/metastatic) cancers including melanoma, squamous and non-squamous NSCLC, cutaneous squamous cell carcinoma, head and neck squamous cell carcinoma, Merkel cell carcinoma, and lung, gastric, and urothelial cancer [
125,
136,
138,
139,
140,
141,
142,
143,
144,
145,
146]. Noteworthily, clinical use of ICI showed that, among the most responsive cancers, there is a subset of tumors characterized by microsatellite instability/DNA mismatch repair deficiency. These tumors display a high number of somatic mutations, leading to the expression of several neo-epitopes/neoantigens [
147]. This association between clinical benefit and tumor mutational burden (TMB) was first shown with ipilimumab (anti-CTLA-4) in advanced melanoma patients [
148,
149]. Mismatch repair deficiency with consequently high TMB was shown to predict the response of colon cancer and later on of other solid tumors to PD-1 blockade [
150].
7. Synergy between Radiotherapy and ICI
Local tumor irradiation has the potential to generate an immune response against the targeted tumor. Such a response would also be expected to act on metastatic lesions that share antigenic characteristics with the original tumor, providing protection even toward not yet diagnosed secondary lesions. Conversely, irradiation of primary lesions alone does not usually elicit an effective potent antitumor immune response: local recurrences are frequent, and immune-mediated regression of distant tumors (abscopal effect) is very rare [
157].
Effectiveness of ICI therapy relies on the ability of tumor cells to potentially prime an immune response, a feature depending on tumor cell intrinsic characteristics, (induced) TMB, and other TME factors. TAAs might be targeted by the immune system, provided they will be taken up and presented by APCs to T cells in an immunogenic context; T cells are primed and differentiate to effector cells, which can infiltrate tumors and possibly kill cancer cells [
37].
The synergy between local RT and CTLA-4 blockade in poorly immunogenic tumors was shown in mouse models of mammary and colon carcinoma where single therapies were not effective. CTLA-4 blockade could induce an abscopal effect on metastatic lesions, when primary tumors were locally irradiated, with the effect showing a correlation with the frequency of tumor-specific IFNγ-secreting CD8 T cells [
159,
160]. Remarkably, the abscopal effects and the activation of tumor-specific T cells were more evident when the radiation dose was hypo-fractionated compared with a single high dose or a higher number of lower fractions [
159]. From a mechanistic point of view, the synergy between RT and CTLA-4 blockade results in the expansion of TAA-specific CD8 TILs, with the RT broadening the TCR repertoire and the anti-CTLA-4 mAb promoting activation and expansion of these selected T-cell clones [
162]. Experimental models have shown that RT combined with ICI targeting PD-1/PD-L1 improved survival in mice with melanoma, breast cancer, NSCLC, and glioma [
163,
164,
165].
8. Synergy between PARPi and ICI
By compromising HR in tumor cells, PARPis can generate unrecoverable DNA damage, leading to increased TMB and generation of neoantigens. The generation of potentially immunogenic neoantigens correlates with better prognosis, as already mentioned above, and can synergize with ICI, improving the therapeutic response [
34,
108,
109,
110,
148,
181,
182].
PARP inhibition affects the TME. By boosting the cGAS/STING pathway, PARPis sustain inflammation and the secretion of IFN-I and several other cytokines and chemokines, resulting in recruitment of immune cells, including TAA-specific CD8 T cells. These effects could be further enhanced by ICI and are particularly relevant in those tumors otherwise cold from the immune infiltration point of view [
183]. Using a BRCA1-deficient ovarian cancer mouse model, PARPi was shown to increase the therapeutic effects of CTLA-4 blockade which, as a single therapy, had limited benefit. The therapeutic effect was dependent on T-cell responses and generated a protective immunological memory.
Several clinical trials in phase I/II evaluated the association of PARPis and ICI (targeting CTLA-4, PD-1, or PD-L1) in triple-negative breast, ovarian, and prostate cancers. Some of these trials are still ongoing, whereas other have already published (partial) results. The combination of olaparib and tremelimumab (anti-CTLA-4) was tolerable in recurrent BRCA-associated ovarian cancer, with preliminary results showing evidence of therapeutic effect [
188]. Combinations of PARPis (olaparib, pamiparib, and niraparib) and anti-PD-L1 (durvalumab) and of PARPis and anti-PD-1 (pembrolizumab and tislelizumab) were also shown to be well tolerated and associated in some cases, with a certain clinical benefit [
189,
190,
191]. Olaparib and atezolimumab (anti-PD-L1) increased IFNγ, TNFα, and CXCL9/CXCL10 expression and tumor infiltration by lymphocytes. Although the clinical activity in recurrent ovarian cancer was modest, the increased IFNγ production was associated with improved progression-free survival [
192].
9. Perspectives: triple RT, PARPi and ICI combinations to overcome respective limitations
As described above RT, PARPis and ICI have a certain therapeutic success when used alone but it is their combination that can result in a better and prolonged disease control. RT and PARPis synergize in inducing DNA damage and tumor cell death. They also induce immune stimulating factors potentially generating an immunogenic microenvironment and favoring immune infiltration. However, they also activate immune suppressive mechanisms and indeed the induction of a systemic immune response with abscopal effects remains uncommon and/or limited. On the other hand, ICIs can lower the threshold for immune activation, reinvigorate exhausted T cells, and dampen the action of regulatory T cells, consequently sustaining systemic immune responses and abscopal effect. Nevertheless, to be effective ICIs require a TME that allows priming of immune responses to tumor-associated antigens and tumor infiltration by leukocytes. Combinations of immunotherapy with therapies that favor priming of immune responses, such as RT, obtained important therapeutic success in clinical studies, with protocols including different forms of RT and ICI having been approved for several (advanced) cancers. Also the more recent association of PARPis and ICIs showed some clinical benefits. Altogether these results and the considerations expressed above encourage the use of combined therapies that include RT, PARPis and ICIs.
Promising results from initial studies in experimental models confirmed that the triple combination of RT, PARPis and ICI improve tumor infiltrate, and prime and unleash anti-tumor, T-cell-mediated, immune responses in mouse models [112][194]. Several phase I to III clinical trials, aimed at exploring different combinations of radiotherapy, PARPis and ICIs, included at least one arm with the concomitant or sequential use of these three therapeutic agents (often in addition to standard chemo-therapy). The effects of PARPis together with RT and ICI, targeting either CTLA-4 or PD-1/PD-L1 or both pathways, will be assessed in NSCLC, SCLC, breast, prostate, pancreatic, gastroesophageal, rectal, head and neck carcinomas. Many of these trials are still recruiting or not yet active. A wealthy of results will be available on these promising therapeutic combinations in forthcoming years [see Table 1 in Rosado et al, Cancers 2023, 15(4), 1093].