Amyotrophic Lateral Sclerosis is a progressive neurodegenerative disease and is the most common adult motor neuron disease. The disease pathogenesis is complex with the perturbation of multiple pathways proposed, including mitochondrial dysfunction, RNA processing, glutamate excitotoxicity, endoplasmic reticulum stress, protein homeostasis and endosomal transport/extracellular vesicle (EV) secretion. EVs are nanoscopic membrane-bound particles that are released from cells, involved in the intercellular communication of proteins, lipids and genetic material, and there is increasing evidence of their role in amyotrophic lateral sclerosis (ALS).
- amyotrophic lateral sclerosis
- exosomes
- extracellular vesicles
1. Extracellular Vesicles in Amyotrophic Lateral Sclerosis
1.1. Amyotrophic Lateral Sclerosis Associated Genes Involved in Vesicular Pathways
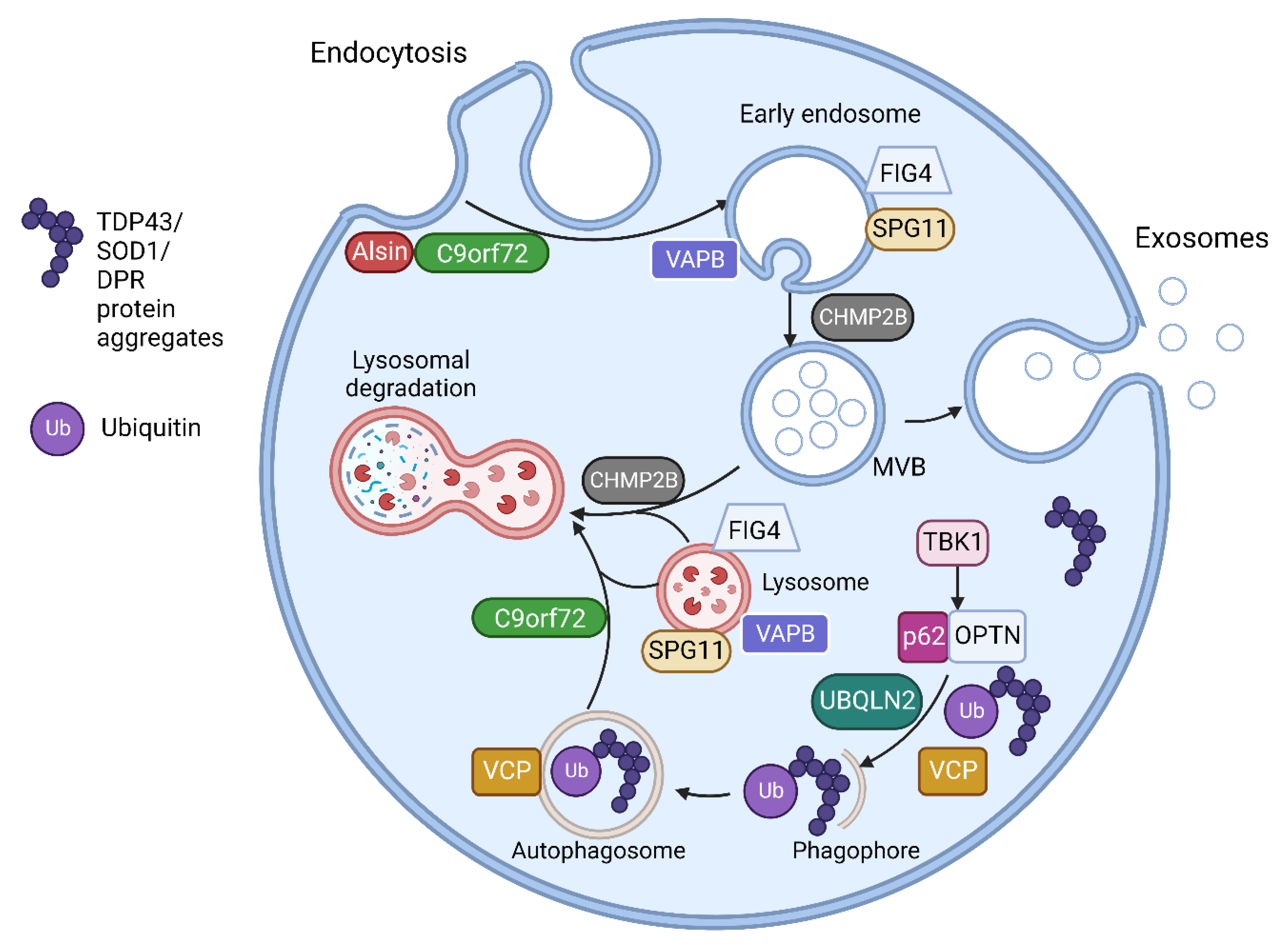
| Gene | Proteins | Molecular Pathways Affected |
|---|---|---|
| C9orf72 [17][26] | C9orf72 short and long isoforms | Loss of function in vesicle trafficking, autophagy and endo-lysosomal pathway Gain of toxicity with development of RNA foci and DPR |
| VAPB [20][21] | Vesicle-associated membrane protein-associated protein B/C | Aggregation of VAPB protein, altered autophagy and vesicular transport, accumulation of RBPs |
| FIG4 [18] | Polyphosphoinositide phosphatase | Loss of function in trafficking of endosomal vesicles to golgi and autophagy regulation |
| ALS2 [19] | Alsin | Alteration of Rab5-mediated pathway with dysregulation of endosomal trafficking Altered trafficking of AMPA receptors causing glutamate toxicity |
| CHMP2B [16] | Charged multivesicular body protein 2b |
Dysfunction of autophagy and endo-lysosomal pathway, resulting in accumulation of enlarged endosomes and autophagic organelles |
| SPG11 [22] | Spatacsin | Impaired autophagy, lipid sorting in late endosomes and lysosomal dysfunction with lipid accumulation |
| SQSTM1 [27] | Sequestosome-1/p62 | Dysfunction of autophagy and protein degradation through UPS |
| OPTN [28] | Optineurin | Golgi fragmentation, impaired autophagy and vesicular transport Loss of inhibitory action on NF-κB leading to abnormal inflammatory response |
| UBQLN2 [29] | Ubiquilin 2 | Impaired protein degradation via UPS and dysfunction of autophagy and endo-lysosomal pathway |
| VCP [30][31] | Valosin Containing Protein | Impaired protein degradation via UPS and dysfunction of autophagy and endo-lysosomal pathway |
| TBK1 [32] | Tank Binding Kinase 1 | Dysregulation of multiple autophagy pathways |
1.2. Extracellular Vesicle Mediated Transfer of Misfolded Proteins and miRNAs in Amyotrophic Lateral Sclerosis
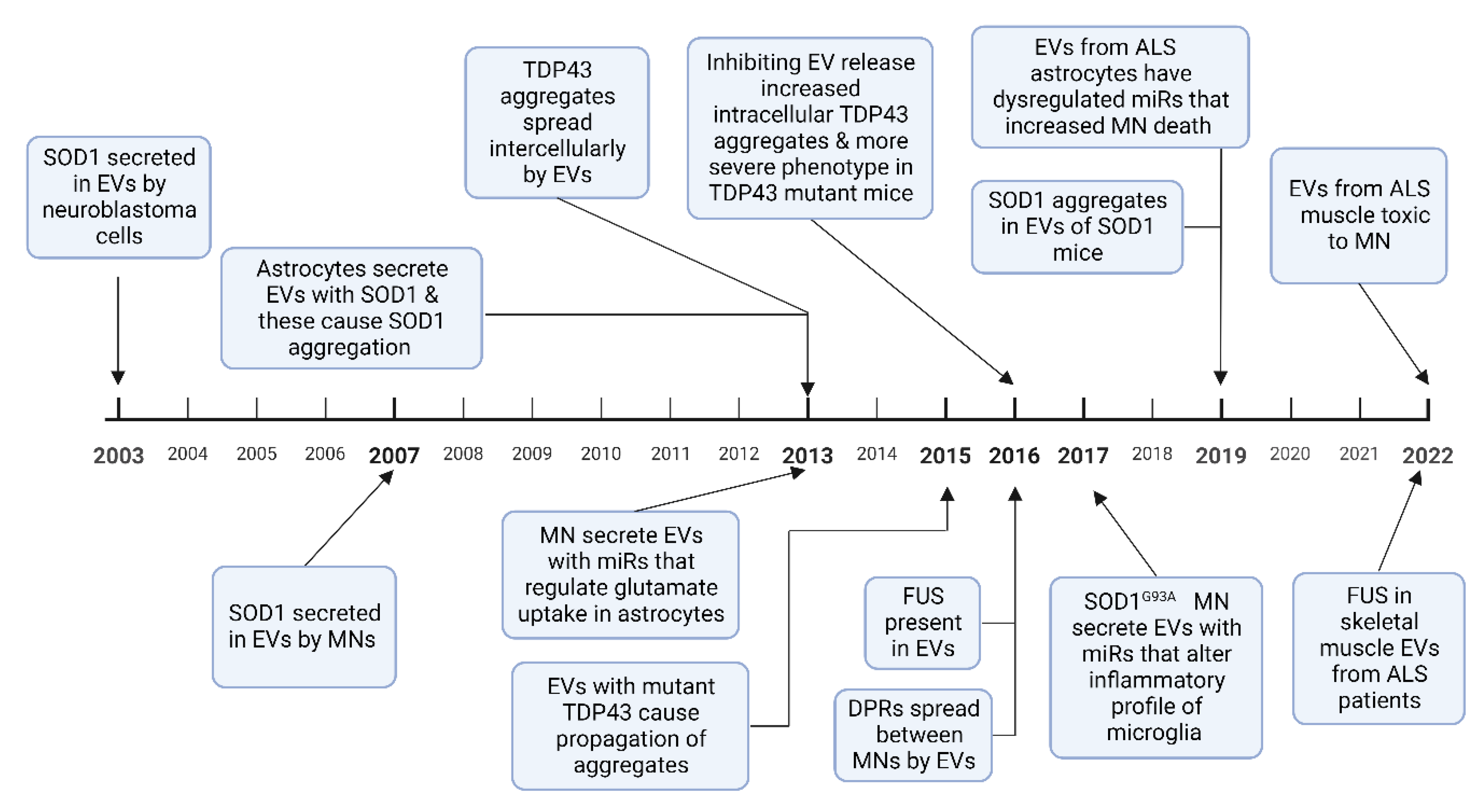
1.2.1. SOD1
1.2.2. TDP 43
1.2.3. FUS
1.2.4. Dipeptide Repeat Proteins
1.2.5. RNA Transport by EVs
2. Therapeutic Application of Extracellular Vesicles in Amyotrophic Lateral Sclerosis
This entry is adapted from the peer-reviewed paper 10.3390/life13010121
References
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. Int. J. Mol. Sci. 2020, 21, 4407.
- Pascua-Maestro, R.; González, E.; Lillo, C.; Ganfornina, M.D.; Falcón-Pérez, J.M.; Sanchez, D. Extracellular Vesicles Secreted by As-troglial Cells Transport Apolipoprotein D to Neurons and Mediate Neuronal Survival Upon Oxidative Stress. Front. Cell Neurosci. 2019, 12, 526.
- Korkut, C.; Li, Y.; Koles, K.; Brewer, C.; Ashley, J.; Yoshihara, M.; Budnik, V. Regulation of Postsynaptic Retrograde Signaling by Presynaptic Exosome Release. Neuron 2013, 77, 1039–1046.
- Xu, B.; Zhang, Y.; Du, X.-F.; Li, J.; Zi, H.-X.; Bu, J.-W.; Yan, Y.; Han, H.; Du, J.-L. Neurons secrete miR-132-containing exosomes to regulate brain vascular integrity. Cell Res. 2017, 27, 882–897.
- Bahram Sangani, N.; Gomes, A.R.; Curfs, L.M.G.; Reutelingsperger, C.P. The role of Extracellular Vesicles during CNS development. Prog. Neurobiol. 2021, 205, 102124.
- Marostica, G.; Gelibter, S.; Gironi, M.; Nigro, A.; Furlan, R. Extracellular Vesicles in Neuroinflammation. Front. Cell Dev. Biol. 2021, 8, 623039.
- Thompson, A.G.; Gray, E.; Heman-Ackah, S.M.; Mäger, I.; Talbot, K.; El Andaloussi, S.; Wood, M.J.; Turner, M.R. Extracellular vesicles in neurodegenerative disease—Pathogenesis to biomarkers. Nat. Rev. Neurol. 2016, 12, 346–357.
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310.
- Grassano, M.; Calvo, A.; Moglia, C.; Sbaiz, L.; Brunetti, M.; Barberis, M.; Casale, F.; Manera, U.; Vasta, R.; Canosa, A.; et al. Systematic evaluation of genetic mutations in ALS: A population-based study. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1190–1193.
- McCluskey, G.; Duddy, W.; Haffey, S.; Morrison, K.; Donaghy, C.; Duguez, S. Epidemiology and survival trends of motor neurone disease in Northern Ireland from 2015 to 2019. Eur. J. Neurol. 2021, 29, 707–714.
- Shepheard, S.R.; Parker, M.D.; Cooper-Knock, J.; Verber, N.S.; Tuddenham, L.; Heath, P.; Beauchamp, N.; Place, E.; Sollars, E.S.; Turner, M.R.; et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 510–518.
- Liu, X.; He, J.; Gao, F.-B.; Gitler, A.D.; Fan, D. The epidemiology and genetics of Amyotrophic lateral sclerosis in China. Brain Res. 2018, 1693, 121–126.
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of amyotrophic lateral sclerosis: Seeking therapeutic targets in the era of gene therapy. J. Hum. Genet. 2022, 2022, 1–22.
- Gonçalves, J.P.N.; Leoni, T.B.; Martins, M.P.; Peluzzo, T.M.; Dourado, M.E.T., Jr.; Saute, J.A.M.; Covaleski, A.P.P.M.; de Oliveira, A.S.B.; Claudino, R.; Marques, W., Jr.; et al. Genetic epidemiology of familial ALS in Brazil. Neurobiol. Aging 2021, 102, 227-e1–227-e4.
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206.
- Ugbode, C.; West, R.J. Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 147, 105144.
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447.
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88.
- Miceli, M.; Exertier, C.; Cavaglià, M.; Gugole, E.; Boccardo, M.; Casaluci, R.R.; Ceccarelli, N.; De Maio, A.; Vallone, B.; Deriu, M.A. ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules. Biology 2022, 11, 77.
- Tripathi, P.; Guo, H.; Dreser, A.; Yamoah, A.; Sechi, A.; Jesse, C.M.; Katona, I.; Doukas, P.; Nikolin, S.; Ernst, S.; et al. Pathomechanisms of ALS8: Altered autophagy and defective RNA binding protein (RBP) homeostasis due to the VAPB P56S mutation. Cell Death Dis. 2021, 12, 466.
- Mao, D.; Lin, G.; Tepe, B.; Zuo, Z.; Tan, K.L.; Senturk, M.; Zhang, S.; Arenkiel, B.R.; Sardiello, M.; Bellen, H.J. VAMP associated proteins are required for autophagic and lysosomal degradation by promoting a PtdIns4P-mediated endosomal pathway. Autophagy 2019, 15, 1214–1233.
- Branchu, J.; Boutry, M.; Sourd, L.; Depp, M.; Leone, C.; Corriger, A.; Vallucci, M.; Esteves, T.; Matusiak, R.; Dumont, M.; et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol. Dis. 2017, 102, 21–37.
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. 2016, 113, 4039–4044.
- Zheng, J.; Tan, J.; Miao, Y.-Y.; Zhang, Q. Extracellular vesicles degradation pathway based autophagy lysosome pathway. Am. J. Transl. Res. 2019, 11, 1170–1183.
- Wei, W.; Pan, Y.; Yang, X.; Chen, Z.; Heng, Y.; Yang, B.; Pu, M.; Zuo, J.; Lai, Z.; Tang, Y.; et al. The Emerging Role of the Interaction of Extracellular Vesicle and Au-tophagy-Novel Insights into Neurological Disorders. J. Inflamm. Res. 2022, 15, 3395–3407.
- Pang, W.; Hu, F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2020, 157, 334–350.
- Davidson, J.M.; Chung, R.S.; Lee, A. The converging roles of sequestosome-1/p62 in the molecular pathways of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Neurobiol. Dis. 2022, 166, 105653.
- Markovinovic, A.; Cimbro, R.; Ljutic, T.; Kriz, J.; Rogelj, B.; Munitic, I. Optineurin in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog. Neurobiol. 2017, 154, 1–20.
- Osaka, M.; Ito, D.; Yagi, T.; Nihei, Y.; Suzuki, N. Evidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 24, 1617–1629.
- Scarian, E.; Fiamingo, G.; Diamanti, L.; Palmieri, I.; Gagliardi, S.; Pansarasa, O. The Role of VCP Mutations in the Spectrum of Am-yotrophic Lateral Sclerosis-Frontotemporal Dementia. Front. Neurol. 2022, 13, 841394.
- Ferrari, V.; Cristofani, R.; Tedesco, B.; Crippa, V.; Chierichetti, M.; Casarotto, E.; Cozzi, M.; Mina, F.; Piccolella, M.; Galbiati, M.; et al. Valosin Containing Protein (VCP): A Multistep Regulator of Autophagy. Int. J. Mol. Sci. 2022, 23, 1939.
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5.
- Mackenzie, I.R.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434.
- Cicardi, M.E.; Marrone, L.; Azzouz, M.; Trotti, D. Proteostatic imbalance and protein spreading in amyotrophic lateral sclerosis. EMBO J. 2021, 40, e106389.
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyo-trophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 262.
- Kanouchi, T.; Ohkubo, T.; Yokota, T. Can regional spreading of amyotrophic lateral sclerosis motor symptoms be explained by prion-like propagation? J. Neurol. Neurosurg. Psychiatry 2012, 83, 739–745.
- Hartmann, A.; Muth, C.; Dabrowski, O.; Krasemann, S.; Glatzel, M. Exosomes and the Prion Protein: More than One Truth. Front. Neurosci. 2017, 11, 194.
- Weng, S.; Lai, Q.-L.; Wang, J.; Zhuang, L.; Cheng, L.; Mo, Y.; Liu, L.; Zhao, Z.; Zhang, Y.; Qiao, S. The Role of Exosomes as Mediators of Neuroinflammation in the Pathogenesis and Treatment of Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 899944.
- Tsunemi, T.; Ishiguro, Y.; Yoroisaka, A.; Hattori, N. Analysis of α-Synuclein in Exosomes. Methods Mol. Biol. 2021, 2322, 41–45.
- Mondola, P.; Ruggiero, G.; Serù, R.; Damiano, S.; Grimaldi, S.; Garbi, C.; Monda, M.; Greco, D.; Santillo, M. The Cu,Zn superoxide dismutase in neuroblastoma SK-N-BE cells is exported by a microvesicles dependent pathway. Mol. Brain Res. 2003, 110, 45–51.
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007, 428, 43–46.
- Basso, M.; Pozzi, S.; Tortarolo, M.; Fiordaliso, F.; Bisighini, C.; Pasetto, L.; Spaltro, G.; Lidonnici, D.; Gensano, F.; Battaglia, E.; et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: Implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J. Biol. Chem. 2013, 288, 15699–15711.
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like Properties of Pathological TDP-43 Aggregates from Diseased Brains. Cell Rep. 2013, 4, 124–134.
- Morel, L.; Regan, M.; Higashimori, H.; Ng, S.K.; Esau, C.; Vidensky, S.; Rothstein, J.; Yang, Y. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J. Biol. Chem. 2013, 288, 7105–7116.
- Ding, X.; Ma, M.; Teng, J.; Teng, R.K.; Zhou, S.; Yin, J.; Fonkem, E.; Huang, J.H.; Wu, E.; Wang, X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 2015, 6, 24178–24191.
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201.
- Kamelgarn, M.; Chen, J.; Kuang, L.; Arenas, A.; Zhai, J.; Zhu, H.; Gal, J. Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2016, 1862, 2004–2014.
- Westergard, T.; Jensen, B.K.; Wen, X.; Cai, J.; Kropf, E.; Iacovitti, L.; Pasinelli, P.; Trotti, D. Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72 -ALS/FTD. Cell Rep. 2016, 17, 645–652.
- Pinto, S.; Cunha, C.; Barbosa, M.; Vaz, A.R.; Brites, D. Exosomes from NSC-34 Cells Transfected with hSOD1-G93A Are Enriched in miR-124 and Drive Alterations in Microglia Phenotype. Front. Neurosci. 2017, 11, 273.
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.-M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Stacey, R.G.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759.
- Varcianna, A.; Myszczynska, M.A.; Castelli, L.M.; O’Neill, B.; Kim, Y.; Talbot, J.; Nyberg, S.; Nyamali, I.; Heath, P.R.; Stopford, M.J.; et al. Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. Ebiomedicine 2019, 40, 626–635.
- Le Gall, L.; Duddy, W.J.; Martinat, C.; Mariot, V.; Connolly, O.; Milla, V.; Anakor, E.; Ouandaogo, Z.G.; Millecamps, S.; Lainé, J.; et al. Muscle cells of sporadic amyotrophic lateral sclerosis patients secrete neurotoxic vesicles. J. Cachexia Sarcopenia Muscle 2022, 13, 1385–1402.
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625.
- Münch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. 2011, 108, 3548–3553.
- Massenzio, F.; Peña-Altamira, E.; Petralla, S.; Virgili, M.; Zuccheri, G.; Miti, A.; Polazzi, E.; Mengoni, I.; Piffaretti, D.; Monti, B. Microglial overexpression of fALS-linked mutant SOD1 induces SOD1 processing impairment, activation and neurotoxicity and is counteracted by the autophagy inducer trehalose. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3771–3785.
- Turner, B.J.; Atkin, J.; Farg, M.A.; Zang, D.W.; Rembach, A.; Lopes, E.C.; Patch, J.D.; Hill, A.; Cheema, S.S. Impaired Extracellular Secretion of Mutant Superoxide Dismutase 1 Associates with Neurotoxicity in Familial Amyotrophic Lateral Sclerosis. J. Neurosci. 2005, 25, 108–117.
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911.
- Schmitz, A.; Marques, J.P.; Oertig, I.; Maharjan, N.; Saxena, S. Emerging Perspectives on Dipeptide Repeat Proteins in C9ORF72 ALS/FTD. Front. Cell. Neurosci. 2021, 15, 637548.
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606.
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The MicroRNA Spectrum in 12 Body Fluids. Clin. Chem. 2010, 56, 1733–1741.
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659.
- Jovičić, A.; Gitler, A.D. Distinct repertoires of microRNAs present in mouse astrocytes compared to astrocyte-secreted exosomes. PLoS ONE 2017, 12, e0171418.
- Men, Y.; Yelick, J.; Jin, S.; Tian, Y.; Chiang, M.S.R.; Higashimori, H.; Brown, E.; Jarvis, R.; Yang, Y. Exosome reporter mice reveal the involvement of exosomes in mediating neuron to astroglia communication in the CNS. Nat. Commun. 2019, 10, 1–18.
- Otake, K.; Adachi-Tominari, K.; Nagai, H.; Saito, M.; Sano, O.; Hirozane, Y.; Iwata, H. Quantitative comparison of the mRNA content of human iPSC-derived motor neurons and their extracellular vesicles. FEBS Open Bio 2021, 11, 494–506.
- Garbuzova-Davis, S.; Sadanandan, N.; Lee, J.-Y. Extracellular vesicle-based therapy for amyotrophic lateral sclerosis. Brain Circ. 2021, 7, 23–28.
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759.
- Gagliardi, D.; Bresolin, N.; Comi, G.P.; Corti, S. Extracellular vesicles and amyotrophic lateral sclerosis: From misfolded protein vehicles to promising clinical biomarkers. Cell. Mol. Life Sci. 2020, 78, 561–572.
- Kalani, A.; Tyagi, N. Exosomes in neurological disease, neuroprotection, repair and therapeutics: Problems and perspectives. Neural Regen. Res. 2015, 10, 1565–1567.
- Cooper, J.M.; Wiklander, P.B.O.; Nordin, J.Z.; Al-Shawi, R.; Wood, M.J.; Vithlani, M.; Schapira, A.H.V.; Simons, J.P.; El-Andaloussi, S.; Alvarez-Erviti, L. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 2014, 29, 1476–1485.
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345.
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Mesenchymal Stem Cells: A Potential Therapeutic Approach for Amyotrophic Lateral Sclerosis? Stem Cells Int. 2019, 2019, 3675627.
- Ciervo, Y.; Ning, K.; Jun, X.; Shaw, P.J.; Mead, R.J. Advances, challenges and future directions for stem cell therapy in amyotrophic lateral sclerosis. Mol. Neurodegener. 2017, 12, 85.
- Araldi, R.P.; D’Amelio, F.; Vigerelli, H.; De Melo, T.C.; Kerkis, I. Stem Cell-Derived Exosomes as Therapeutic Approach for Neurodegenerative Disorders: From Biology to Biotechnology. Cells 2020, 9, 2663.
- Lee, M.; Ban, J.-J.; Kim, K.Y.; Jeon, G.S.; Im, W.; Sung, J.-J.; Kim, M. Adipose-derived stem cell exosomes alleviate pathology of amyotrophic lateral sclerosis in vitro. Biochem. Biophys. Res. Commun. 2016, 479, 434–439.
- Bonafede, R.; Scambi, I.; Peroni, D.; Potrich, V.; Boschi, F.; Benati, D.; Bonetti, B.; Mariotti, R. Exosome derived from murine adipose-derived stromal cells: Neuroprotective effect on in vitro model of amyotrophic lateral sclerosis. Exp. Cell Res. 2016, 340, 150–158.
- Calabria, E.; Scambi, I.; Bonafede, R.; Schiaffino, L.; Peroni, D.; Potrich, V.; Capelli, C.; Schena, F.; Mariotti, R. ASCs-Exosomes Recover Coupling Efficiency and Mitochondrial Membrane Potential in an in vitro Model of ALS. Front. Neurosci. 2019, 13, 1070.
- Bonafede, R.; Brandi, J.; Manfredi, M.; Scambi, I.; Schiaffino, L.; Merigo, F.; Turano, E.; Bonetti, B.; Marengo, E.; Cecconi, D.; et al. The Anti-Apoptotic Effect of ASC-Exosomes in an In Vitro ALS Model and Their Proteomic Analysis. Cells 2019, 8, 1087.
- Bonafede, R.; Turano, E.; Scambi, I.; Busato, A.; Bontempi, P.; Virla, F.; Schiaffino, L.; Marzola, P.; Bonetti, B.; Mariotti, R. ASC-Exosomes Ameliorate the Disease Progression in SOD1(G93A) Murine Model Underlining Their Potential Therapeutic Use in Human ALS. Int. J. Mol. Sci. 2020, 21, 3651.
- Garbuzova-Davis, S.; Willing, A.E.; Ehrhart, J.; Wang, L.; Sanberg, P.R.; Borlongan, C.V. Cell-Free Extracellular Vesicles Derived from Human Bone Marrow Endothelial Progenitor Cells as Potential Therapeutics for Microvascular Endothelium Restoration in ALS. NeuroMolecular Med. 2020, 22, 503–516.
- Giunti, D.; Marini, C.; Parodi, B.; Usai, C.; Milanese, M.; Bonanno, G.; de Rosbo, N.K.; Uccelli, A. Role of miRNAs shuttled by mesenchymal stem cell-derived small extracellular vesicles in modulating neuroinflammation. Sci. Rep. 2021, 11, 1740.